
Abstract
Cluster of differentiation 24 (CD24) is a cell surface glycoprotein, which is largely present on hematopoietic cells and many types of solid tumor cells. CD24 is known to be involved in a wide range of downstream signaling pathways and neural development, yet the underlying mechanisms are poorly understood. Moreover, its production correlates with poor cancer prognosis, and targeting of CD24 with different antibodies has been shown to inhibit disease progression. Nucleic acid aptamers are oligonucleotides that are selected from random DNA or RNA libraries for high affinity and specific binding to a certain target. Thus, they can be used as an alternative to antibodies. To gain an insight on CD24 role and its interaction partners, we performed several SELEX (systematic evolution of ligands by exponential enrichment) experiments to select CD24-specfiic DNA aptamers. We found that the cell-SELEX approach was the most useful and that using HT-29 cell line presenting CD24 along with CD24 knockdown HT-29 cells has selected six aptamers. For the selected aptamers, we determined dissociation constants in the nanomolar range (18–709 nM) using flow cytometry. These aptamers can be applied as diagnostic tools to track cancer progression and bear a potential for therapeutic use for inhibiting signaling pathways that promote the metastatic process.
Introduction
H
Ser and Thr represent 50% of the amino acid residues in the protein, which can be O-glycosylated with glycans. Four Asp can serve as N-glycosylation sites. This extensive and variable glycosylation pattern shows that post-translational modifications of this molecule can change its recognition properties [7].
Physiologically, CD24 is produced mainly on premature lymphocytes, epithelial, platelet, and neural cells [1,3,8]. During hematopoiesis process, CD24 plays a crucial role in selection and maturation of the different blood cells [1,9]. It serves as a lymphoid differentiation marker and was already identified by Zhou et al. and Wang et al. as a genetic modifier of risk and progression in multiple sclerosis and systemic lupus erythematosus [10,11]. CD24 interaction with sialic acid-binding protein (Siglec-5), which is presented on immune cells, might have an essential role in immune regulation [12,13].
The amount of CD24 in epithelial tissue (17%) is significantly increasing (91%) during tumorigenesis [14]. And it is overpresented on the surface of the majority of solid tumors, including brain, breast, colon, liver, lung, ovary, and prostate carcinomas, implying its role as a prognostic biomarker [15–26]. In patient metastases, the amount of CD24 is even higher than in primary tumors [21,22,27]. A growing number of publications suggest that CD24 production can serve in vitro and in vivo as a predictor of tumor aggressiveness [15–26]. This is in agreement with the results of a CD24 meta-analysis from Lee et al. This study, carried out between 1990 and 2009, showed that higher levels of CD24 were associated not only with malignant transformation of the cells but also with the advanced stage of the cancer progression.
Moreover, CD24 was the most overproduced transcript in highly metastatic cells [28]. For example, pancreatic cancer cells with the CD44+CD24+ESA+ phenotype have a 100-fold increased tumorigenic potential compared with cancer cells, which are not producing these three markers [27]. In many cancers, CD24 is overproduced already at early stages of carcinogenesis. For example, its production correlates with p53 mutation and can be used as an early tumor marker in hepatocellular carcinoma [14,20]. Upregulation of CD24 has also been shown in chemoresistant liver tumors [29]. Together, these findings suggest that CD24 is a worthwhile target in early detection and treatment of cancer, especially in antimetastatic therapy [22].
CD24 has been successfully targeted with antibodies, to identify subpopulations of mammary tumor cells and for sorting of cancer stem cells [23,30,31]. Moreover, it was possible to inhibit the growth of cancer cells up to 90%, in vitro and in vivo, when exposed to anti-CD24 monoclonal antibodies or applying RNA interference. These results were reproduced in four different cell lines with three different anti-CD24 antibodies [14]. Additionally, the inhibition was dose- and time-dependent and corresponded to CD24 production levels of the cells. Inhibition led to CD24 downregulation through lysosomal degradation [32,33]. In clinical trials, CD24 antibodies were successfully used in treatment of B cell lymphoproliferative disease and caused a complete remission in the majority of patients [34,35]. In bladder cancer, the suppression of CD24 significantly reduced lung metastases and led to prolonged survival [22].
Despite all progress in CD24 research, the key interaction partners of CD24 leading to more aggressive course of cancer are still unknown. CD24 was previously reported to interact with P- and E-selectins, fibronectin, collagens I and IV, and laminin [8,36–38]. On one hand, interaction with one of the selectins can play an important role in marginal adhesion and migration of cells under shear forces in the blood stream [36,39]. Thus, CD24-presenting cells might disseminate into the bloodstream more easily. On the other hand, some studies showed that cells are also able to bind to P-selectin in the absence of CD24 and that CD24 production does not increase their ability to bind to P-selectin [38]. For further research as well as for diagnostics and therapeutics, specific binders with high affinity are desirable.
Aptamers represent an interesting alternative to antibodies [40]. The disadvantages of antibodies are their high sensitivity toward pH and temperature, batch-to-batch variability, and immunogenicity. Aptamers are not only cheaper than antibodies but also easier to store, ship, and handle. These short oligonucleotides (mostly <100 nt), which can recognize and bind to their targets, are selected from combinatorial libraries by a process of in vitro evolution, termed SELEX (systematic evolution of ligands by exponential enrichment) [41]. Within the SELEX process, aptamers with unique binding properties can be identified from a large library of oligonucleotides within several iterative cycles.
One selection round contains the following steps: interaction with the target molecule, separation of bound from unbound oligonucleotides, elution, and amplification of the binding aptamers for further selection rounds. These oligonucleotides can serve as binders, inhibitors, activators, sensors, and even as a nucleation point for mineralization [42–46]. Aptamers, which can identify cells presenting CD24, could be used in diagnostics or as an alternative to the antibody-based immunotherapy.
In this article, we report the selection of CD24 aptamers of high specificity and affinity. For the aptamer selection process, we used CD24 presenting HT-29 cells. The corresponding knockdown cell line (HT-29 kdCD24) was used in the negative selection steps. To avoid possible receptor internalization upon oligonucleotide binding, we performed all selection steps at 4°C. The resulting aptamers provide suitable tools to identify CD24 interaction partners and to understand the invasiveness of CD24-positive cells, which might not be influenced by P-selectin binding, as previously assumed [47].
Materials and Methods
Materials
Chemicals
All chemicals were purchased from Sigma–Aldrich (Munich, Germany), Carl Roth (Karlsruhe, Germany), or AppliChem (Darmstadt, Germany). Buffers were prepared by using deionized water obtained from a water purification system (Millipore, Billerica, MA).
Oligonucleotides
The commercial DNAs were synthesized, modified, and purified by Life Technologies (Carlsbad, CA) or Sigma–Aldrich. Oligonucleotides, produced by polymerase chain reaction (PCR) or after polynucleotide kinase reaction for radioactive labeling, were purified with DNA purification kit (NucleoSpin® Gel and PCR Clean-up; Macherey-Nagel, Düren, Germany). For polyacrylamide gel electrophoresis (PAGE) purification, the desired DNA band was cut from polyacrylamide (PAA) gel, eluted into diffusion buffer [500 mM ammonium acetate, pH 8.0, 0.1% sodium dodecyl sulfate (SDS, w/w), 1 mM EDTA, 10 mM magnesium acetate], and transferred on silica columns.
Antibodies
Antibodies were purchased from BioLegend (San Diego, CA), SouthernBiotech (Birmingham, AL), or Santa Cruz Biotechnology (Dallas, TX).
Cells
The HT-29 and HT-29 knockdown CD24 cells, which were transfected with vector bearing the sequence coding the siRNA specific for CD24 knockdown, were kindly provided by U. Schumacher (University Medical Center Hamburg-Eppendorf). The cells were cultivated in Roswell Park Memorial Institute (RPMI) medium, supplemented with 1% (v/v) penicillin–streptomycin solution and 10% (v/v) fetal bovine serum (FBS) to 80%–90% confluency for SELEX or binding assays (20 passages maximum). All cultivated cells were treated under sterile conditions using HERAcell 240i (Thermo Fisher Scientific, Waltham, MA).
Methods
Systematic evolution of ligands by exponential enrichment
To select DNA aptamers for CD24, cell-SELEX was performed. A primary DNA library, consisting of a central 40-nt randomized region flanked by two 18 nt consensus regions (ATCCAGAGTGACGCAGCA-N40-TGGACACGGTGGCTTAGT), was subjected to cell-SELEX process [48].
HT-29 cells and HT-29 knockdown for CD24 and oligonucleotides were incubated in 150 mM HEPES (pH 7.4), supplemented with 1 mM CaCl2, 1 mM MgCl2, increasing amounts of NaCl and competitive DNA, and with bovine serum albumin (BSA; for 0.5 up to 6 h) (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/nat). To avoid DNA internalization, all incubation steps were performed at 4°C. The DNA molecules, which did not bind to the HT-29 kdCD24 cells, were used for the subsequent positive selection steps. After each round of cell-SELEX, the DNA pool was eluted by heating of the HT-29 cell–DNA complexes to 96°C for 2 min and centrifugation (16,000g three times for 30 s). After DNA amplification by PCR, single-stranded DNA (ssDNA) for the next round of selection was prepared.
PCR amplification of DNA for subsequent SELEX rounds
To produce DNA pools for each selection step, nucleic acids were first amplified by conventional solution PCR (PCR-Cycler Mastercycler personal; Eppendorf, Hamburg, Germany) as described below. For the DNA pool from each round of selection, the number of PCR cycles was optimized. In the next step, the ssDNAs were created by enzymatic hydrolyzation of the complementary strand.
Generation of ssDNA
To generate single-stranded DNA for the next selection step, reverse strands of the DNA molecules in pool were phosphorylated at the 5′-end during PCR by using modified reverse primer. To create DNA single strands, the PCR product was incubated with λ-Exonuclease (Thermo Fisher Scientific), which removes 5′-phosphorylated mononucleotides in the DNA double strand. The resulting DNA single strands were purified with RNA Clean & Concentrator (Zymo Research, Irvine, CA) and quantified with a NanoDrop (ND 1000; Thermo Fisher Scientific). For the incubation with target, the obtained DNA was denatured and slowly cooled down to 4°C.
Polymerase chain reaction
The PCR amplification protocol included an initial denaturation step at 95°C for 2.5 min, followed by cycles of heating to 95°C for 0.5 min, 56.3°C for 0.5 min, and 72°C for 0.5 min (Table 1). A final extension step was performed for 3 min at 72°C. After PCR (∼2–22 cycles), the products were purified with a DNA purification kit (NucleoSpin Gel and PCR Clean-up) (Tables 2 and 3). DNA amounts were quantified with a NanoDrop (ND 1000).
X, in subsequent rounds, the amount of DNA applied was reduced. After incubation and elution, the resulting DNA was amplified. The DNA amounts are listed in Supplementary Table S1.
PCR, polymerase chain reaction.
Cellular assays
Cells were passaged three times after cryoconservation and tested for the production of CD24 by flow cytometry (FACSCalibur™; BD Biosciences, Franklin Lakes, NJ) before use. Dead cells as well as cell debris and some bigger cells (in apoptotic status or cell lumps) were gated out. The Kd values were calculated using one-site binding model.
Antibody binding
CD24 antibody binding flow cytometry
For antibody binding, the cells were first detached with Accutase or with trypsin (MP Biomedicals, Fisher Scientific, Hampton, NH) for 7 min at 37°C. Cells were pelleted at 500g and resuspended in SELEX buffer or in phosphate buffered saline (PBS). After incubation with 1 μg anti-CD24 antibody (clone ML5) for 30 min at 4°C, the cells were washed four times and incubated with 1 μg of fluorescently labeled goat anti-mouse secondary antibody. Finally, the cells were washed three times and resuspended in PBS. The mean fluorescence was analyzed by flow cytometry. For the measurement of cross-reactivity, cells were incubated only with the secondary antibody.
DNA binding
Pull-down assay
For pull-down tests, the DNA (10 pmol) was radioactively labeled with γ-32P-ATP (10 pmol) using polynucleotide kinase (10 U). After 30 min of incubation at 37°C, the labeled DNA was precipitated with isopropanol washed, PAGE-purified, and dissolved in ddH2O. The purified radiolabeled 32P-DNA (1 μL corresponding to 0.05 pmol) was mixed with scintillation cocktail (1 mL; Ultima Gold from PerkinElmer, Waltham, MA) and quantified with a scintillation counter (TRI-CARB 2500 TR, Packard; PerkinElmer). For the pull-down assay, cells were resuspended in SELEX buffer. 5 × 105 cells were incubated with radioactively labeled DNAs (30,000–80,000 counts per second, maximum 50 nM solutions) under SELEX conditions (30 min at 4°C), subsequently washed four times with SELEX buffer, and resuspended in 100 μL SELEX buffer.
The amount of the radioactivity was quantified in a scintillation counter, and the percentage of radioactivity retained on the cells was calculated.
Cytofluorometric analysis
The DNA for the binding studies was fluorescently labeled (fluorescein isothiocyanate [FITC] or cyanine 5 [Cy5]) by PCR using 5′-labeled forward primers or purchased from Sigma–Aldrich. For the binding studies with DNA pools from SELEX rounds, cells were prepared as for pull-down studies and incubated with DNA solution (500 nM) under SELEX conditions. In the case of Kd measurements, HT-29 cells were incubated with a DNA concentration series from 0.5 to 500 nM or from 32 to 4,000 nM (for weak binders).
The binding affinity of the fluorescently labeled DNAs to the cells was shown graphically. The histograms were estimated as logarithmic functions of the mean fluorescences of cell populations versus cell numbers.
Competition assays
For the competition assay, cells were prepared as described above and incubated on ice for 30 min with Cy5-labeled aptamer solution (136 nM), then 30 min with anti-CD24 antibody and 30 min with the FITC-labeled secondary antibody. The incubation time was adjusted for all samples to 1.5 h at 4°C. Fluorescence shifts were measured at 488 nm (antibody binding) as well as at 647 nm (aptamer binding).
DNA assays
Diversity assay
For the diversity assay, quantitative PCR (qPCR) was performed for DNA pools from each selection round. For the products after 10 PCR cycles, the melting curve was measured and the range of melting points was calculated. Comparison with the starting library pool and with DNA solution of one single DNA species allowed evaluating the SELEX progression. The measured DNA pools, after 10 PCR cycles, were controlled on native PAA (20 min at 250 V).
DNA aptamer stability assay
The stability of different DNAs (40 nM) was assessed in cell culture medium (RPMI) supplemented with 10% FBS at 37°C. Samples of 10 μL were taken at determined time intervals (up to 24 h) and immediately frozen with liquid nitrogen. The samples were loaded with dye-free loading solution, and PAA-electrophoresis (10%) under native conditions was performed (40 min at 250 V). The gel was analyzed with a Gel Doc™ XR+ Gel Documentation System (BioRad, Hercules, CA) and ImageLab
Aptamer structure studies (circular dichroism spectroscopy)
The folding of the aptamer CD24A_2 was assessed by CD spectroscopy. Solutions of 100 μM were prepared in 20 mM HEPES buffer (pH 7.4) containing MgCl2 (1 mM) and CaCl2 (1 mM). As a control, 100 μM DNA solution in ddH2O was used. The DNAs were supplemented with LiCl, KCl, NaCl, or with NaCl plus KCl, 150 mM each (SELEX conditions). After denaturation and slow refolding (cooldown to 4°C), the DNA solutions were kept either on ice or at room temperature. The samples were measured in a CD spectrometer (J-815 C; Jasco, Pfungstadt, Germany) at 4°C or 37°C. For each condition, 30 spectra were collected and a mean spectrum was plotted for each sample.
Results
To select CD24-specific DNA aptamers, we performed cell-SELEX based on cell lines from the same origin. We used HT-29 cells presenting the target and CD24 knockdown HT-29 cells (HT-29 kdCD24) for negative selection. In each round, we increased the selection pressure by shortening the incubation times with HT-29 cells (positive selection) and prolonging incubation times with HT-29 kdCD4 cells (negative selection) as well as by gradually increasing the NaCl concentration up to physiological levels (150 mM).
Moreover, from round to round, a lower amount of target cells and a higher number of knockdown cells were used. And as integrins, especially α6 integrin (CD49f+), are highly presented on the surface of HT-29 cells, we added the specific aptamer integrin α6β4-specific DNA aptamer (IDA) to block the corresponding receptor [43,49]. To also block other potential unspecific DNA binders, we included competitive DNA (salmon sperm) and BSA. Based on the described strategy, we obtained six CD24 aptamers (Supplementary Table S1). The enrichment of the selection pool through successive selection was verified in pull-down assays as well as after derivatization with FITC by flow cytometry (Supplementary Fig. S1).
Diversity assay
We modified the analysis of melting curves to adopt it to the complexity of SELEX pools and developed a fast, simple and cheap assay to evaluate SELEX progression [50,51]. We performed qPCR with DNA pools from all rounds of selection and generated melting curves of the PCR products. The process of specific aptamer selection was accompanied by decreasing the variability of the DNA pool (Supplementary Fig. S2), starting from a random library and ultimately leading to few potential aptamers.
To evaluate the decreasing diversity of the DNA pool, we measured the width of the temperature melting peaks for all selected pools. We extracted the widest melting temperature peak, that is, highest melting temperature range, for the DNA starting library as well as for the pool after the first round of selection and a width reduction in subsequent selection rounds (Fig. 1). As a negative control, we measured one single DNA species (Fig. 1, DNA), which showed the lowest melting curve width, as expected. Our data showed that the most significant reductions in DNA pool diversity appeared after the 2nd and 10th round of selection. The enriched pool after the 12th round of selection was cloned and 59 positive clones were sequenced. Four distinct homologous families were identified (Supplementary Table S2). Representative sequences (Supplementary Table S4) from the different families were selected for further analysis. Moreover, the resulting DNA molecules were analyzed for their stability and similarity of theoretical secondary structures as predicted using Mfold algorithm [52]. Additional DNAs whose sequences were similar to the representative DNA molecules were chosen based on their stability or structural characteristics (eg, G-quadruplexes [GQs]) or because they shared sequences or sequence motifs with the other nucleotides (Supplementary Table S3).

Diversity assay monitoring the SELEX progression. The width of the melting temperature peak was calculated for the starting library (L); negative control (DNA), for example, a DNA with same length as the pools and the DNA pools (R1–R12) from corresponding SELEX rounds. Data are presented as means ± SD (n = 2). SD, standard deviation; SELEX, systematic evolution of ligands by exponential enrichment.
DNA cell binding assays
We performed binding assays for selected potential DNA aptamers. Cells were incubated with radioactively labeled aptamers, and after five washing steps, the percentage of DNA retained on the cell surface was measured (Supplementary Fig. S3). We obtained highest affinity for clones No. 19, 52, 31, and 60. These oligonucleotides, whose sequences were found repeatedly among the 59 clones, showed no or only very low affinity to HT-29 cells (Supplementary Fig. S3 and Supplementary Table S3). Remarkably, the DNA No. 60, which was able to interact with the target cells, exhibited just one base deletion in comparison to the nonbinding DNAs No. 36, 49, and 63 (Supplementary Table S3). The four binders from the binding assay were selected to determine the dissociation constants (Kd). Because derivatization with the fluorescent dye Cy5 did not show any significant effect on the specificity of the aptamers, we employed flow cytometry studies to identify further aptamers and to obtain their Kd values.
For flow cytometry measurements, cells were cultured up to 70%–90% confluency and detached using trypsin or accutase. Treatment with trypsin or accutase had no negative effect on CD24 production, as confirmed by CD24 antibody binding (for accutase treatment, see Supplementary Fig. S1). Only viable, that is, propidium iodide (PI)-negative cells were analyzed for antigen production and aptamer binding. Kd values were calculated using the one-site binding model and ranged from 18 to 709 nM (Table 4). The aptamers with the highest affinity were CD24A_2 and CD24A_5 (Figs. 2 and 3). After derivatization of the aptamer with Cy5 for the flow cytometry measurements, we obtained a Kd value of 18 nM for the CD24A_2 aptamer (Fig. 3). The very low affinity of the aptamer to the HT-29 kdCD24 control cells might be caused by residual CD24 expression.

Binding affinities of the aptamers CD24_2
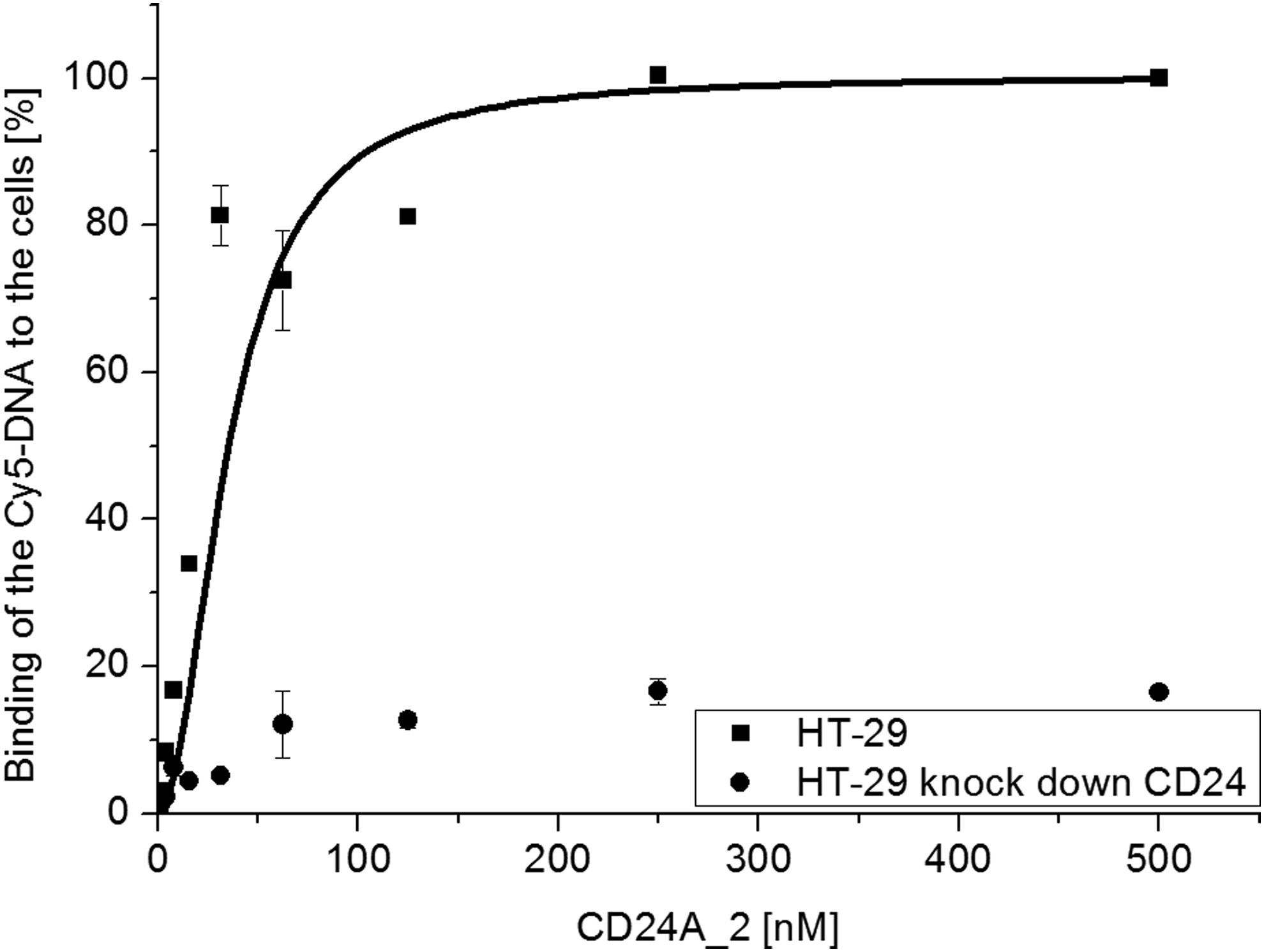
Affinity of CD24A_2 aptamer to HT-29 cells. Increasing amounts of aptamer (0.5–500 nM) were incubated with 5 × 105 HT-29 or CD24 knockdown cells, each. Data represent means ± SD (n = 2).
K
The dissociation constants were obtained using live cells at 4°C. Within the cell-SELEX, six aptamers were selected with Kd values between 18 and 709 nM.
SELEX, systematic evolution of ligands by exponential enrichment.
Characterization of aptamer CD24A_2
The aptamers exhibited similar affinities, both under SELEX conditions (4°C) and at 37°C (Supplementary Fig. S4). However, the staining using aptamer CD24A_2 achieving 8.9-times higher fluorescence signal than the background (HT-29 kdCD24), which is 33% higher than the amount achieved by CD24-specific antibody (clone ML-5; BioLegend). Described antibody showed 6.7-times higher signal over background (HT-29 kdCD24 Supplementary Fig. S5). This indicated that the use of this aptamer leads to better signal-to-noise ratio than the use of the antibody, implying that the aptamer can serve as a potential tool for diagnosis or therapy.
To gain further insights into the binding site at CD24, we performed competition binding assays with the aptamer CD24A_2 versus the ML-5 anti-CD24 antibody, which is known to bind to the nonglycosylated protein core but not to the P-selectin binding site [8]. We saw no competition between CD24A_2 and ML-5 antibody. Thus, it could be possible that CD24A_2 can interact with the P-selectin binding site (Supplementary Fig. S6). Further studies are necessary to evaluate the potential binding site of this aptamer. Note that although ML-5 did not share the same epitope with P-selectin, it was able to stop the process of metastasis [8,14].
Next, we measured the stability of CD24A_2 only in cell culture medium RPMI and in the presence of fetal calf serum (FCS) (Fig. 4 and Supplementary Fig. S7). In RPMI medium, the aptamer showed high stability and 40% of DNA remained intact after 24 h. After 4 h, >80% of aptamers were still intact, whereas the addition of FCS reduced the remaining undigested aptamer to <20% (Fig. 4 and Supplementary Fig. S8).

Stability of the aptamer CD24A_2 in RPMI medium without (dots) or supplemented with 10% fetal bovine serum (squares). The CD24A_2 aptamer was incubated in both media for 0, 1, 2, 5, 10, 15, and 30 min and 1, 2, 4, 8, and 24 h, respectively. The DNA amounts in the samples were detected at 647 nm after electrophoresis and quantified with the software ImageLab™ (see DNA Aptamer Stability Assay section). Data represent means ± SD (n = 2).
Secondary structures predicted by the Mfold software showed similar folding and relatively high stabilities (ΔG = −35 kJ/mol) for the two best binders CD24A_2 and CD24A_5. For CD24A_5 (seven structures at 4°C), three more structures were predicated than for CD24A_2 [52]. Also the prediction at 37°C led to highly stable structures (ΔG = −23 kJ/mol). One of the structures was predicted for both aptamers (CD24A_2 and CD24A_5) at both 37°C and 4°C (Supplementary Fig. S9). The Mfold algorithm predicted other loops, including almost exclusively the nucleotides from constant regions (Supplementary Fig. S9, small letters). Those loops did not seem to have a significant influence on the interaction with CD24 on the HT-29 surface.
Since the selected CD24A_2 aptamer contained high numbers of guanin residues, we used QGRS Mapper (Quadruplex forming G-Rich Sequences) to predict GQ formation [53]. The high probability for GQ formation could be deduced from the G-score value of 21 [54] (Table 5). To verify the predictions, we performed CD spectroscopy in SELEX buffer supplemented with different ions (LiCl, KCl, NaCl, and mixtures thereof) and deionized water. We collected CD spectra in the range from 200 to 320 nm and compared the resulting curves at 4°C and 37°C (Supplementary Fig. S10). The ellipticities (mdeg) of 30 measured spectra were calculated and plotted against the corresponding wavelengths (Fig. 5).
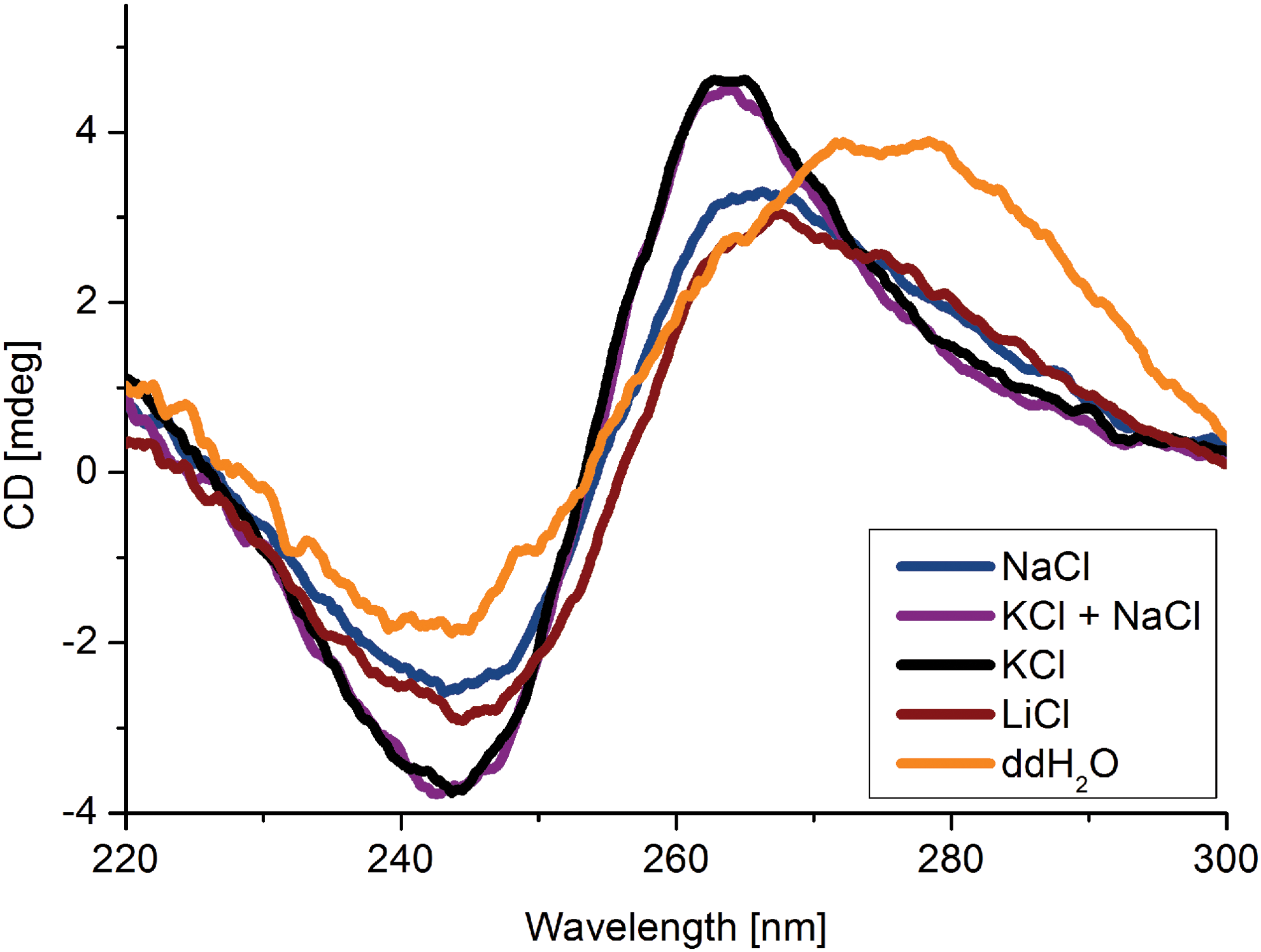
CD measurement of the CD24A_2 aptamer in SELEX medium, in pure water without, and in presence of different metal ions. Spectra and G-quadruplex formation markedly differ from each other depending on the presence of 150 mM monovalent ions (Na+, K+, and Li+). The measured solutions each also contained 1 mM Mg2+ and 1 mM Ca2+ in HEPES buffer pH 7.4. Curves are an average of 25 measurements blanked with the buffers. Color images available online at www.liebertpub.com/nat
The eight guanines, which might form a planar structure, are in bold and underlined. Next to the GQ shown, seven further combinations were predicted, which yielded GQs with G-scores of 19–20.
GQ, G-quadruplex.
At 4°C, the CD spectra of the CD24A_2 aptamer showed two positive maxima at 220 nm and 265 nm and one minimum at 245 nm. These characteristics support the hypothesis that CD24A_2 forms a parallel GQ [54]. This was the case under SELEX conditions (Fig. 5; blue); however, the structure could be further stabilized by potassium ions (Fig. 5; green and black). Lithium ions (Fig. 5; red) exhibited a light destabilizing effect on GQ formation. We also measured the temperature dependence of GQ formation (Supplementary Fig. S10) and observed only a slight destabilization of the structure by increasing the temperature to 37°C, indicating a high stability of the aptamer structure. This might explain why the aptamer bound the target with similar affinity at 4°C and 37°C (Supplementary Fig. S4).
In summary, by applying the cell-SELEX strategy, we selected six aptamers (CD24A_2, _5, _19, _31, _52, and _60, respectively) highly specific for human CD24 presenting HT-29 cells with binding constants in the nanomolar range. We characterized the CD24A_2 aptamer in more detail and determined a Kd value of about 19 nM, at both temperatures 4°C and 37°C (Table 4 and Supplementary Fig. S11). Furthermore, we showed that CD24A_2 had high propensity to form GQ most likely responsible for the high aptamer stability under physiological conditions.
Discussion
To select aptamers for the CD24 molecule, we preferred whole cells as targets instead of targeting solely recombinant protein, as the latter might not represent the accurately folded and/or modified variant of the surface marker. We selected six CD24-specific aptamers using cell-SELEX targeting CD24 presenting colorectal cancer cells (HT-29) after incubation with CD24 knockdown cells (HT-29 kdCD24), for counterselection, in a cell culture-based approach. To the best of our knowledge, the DNA aptamers described here are the first developed to specifically target the CD24 receptor. The successful cell-SELEX method was performed at 4°C to create a constant target level on the cells' surfaces and to avoid endocytotic nucleic acid internalization.
The binding studies show that these aptamers have a high affinity and distinguish between CD24 bearing cells and cells where the production of this molecule was significantly decreased by short hairpin RNA knockdown (Supplementary Fig. S12). The aptamer candidates CD24A_2 and CD24A_5 show the highest affinity to the target cells (Kd = 18 nM, Table 4) at 4°C as well as at 37°C (Supplementary Fig. S11). Structural prediction identifies a stable stem-loop region (ΔG = −23 kJ/mol), which might be proposed as binding motif [52]. The CD24A_2 and CD24A_5 aptamers differed only by one base substitution, which has no significant influence on the affinity to the target cells (Fig. 2). We assume that this nucleobase is not involved in the interaction with the target and does not interfere with folding into the purposeful tertiary structure.
In contrast, the CD24A_60 aptamer recognizes the target while other similar oligonucleotides do not (DNA No. 36, 49 and 63), most likely because of the single base depletion (Supplementary Fig. S3).
A GQ structure of the CD24A_2 aptamer, as predicted by QGRS mapper, is also confirmed by CD spectroscopic analyses (Fig. 5 and Supplementary Fig. S10) [53]. CD24A_2 forms stable GQ in presence of monovalent cations, even also with Li+, differently as it was observed for other aptamers. For example, the GQ formation of AIR-3A aptamer was inhibited by lithium ions (measured up to 100 mM) [54]. Because of the stability and conformational homogeneity of GQ structure of the CD24A_2 aptamer (under different ionic conditions and temperatures), it can be used in different buffer system. This facilitates its further applications.
The GQ of the CD24 aptamer in lithium ions supplemented buffer formed in almost the same manner as in sodium supplemented one. Because of this, we cannot differentiate whether lithium had a destabilizing or a rather neutral influence on the GQ formation. Similar influence of the Li+ on the GQ structures was observed by two different groups before [55,56]. Sodium and lithium might be too small to stabilize the structure of the aptamer in the same manner as potassium does. But solely the ionic radius cannot be responsible for the molecule's folding, like in the case of sodium and calcium with very similar ionic radii of 0.95 and 0.99 Å, respectively. Divalent cations do not promote the formation of GQ structures [55].
The competition study results indicate that the CD24A_2 aptamer does not compete with the CD24-specific antibody ML-5 for binding. This leaves the question concerning the aptamer binding site open. In former studies, all tested CD24-specific antibodies showed inhibition of the cancer progression. This effect was independent of the antibody binding sites.
In this study, we present a very simple, cheap, and quick test to monitor SELEX progression: the diversity assay. It uses the variations in melting point mean values of the DNA pools after each selection round to capture the diversity of molecules present in the corresponding pools. In our case, the biggest decline in diversity occurred after the first and ninth cell-SELEX cycle. In the ninth round, the integrin-specific aptamer IDA was additionally used to block CD49f as potential disruptive target as the presentation of CD24 and CD49f correlates and during the cell-SELEX also integrin binders could have been selected.
After further characterizations, CD24 aptamers might bring us further in research concerning the biochemistry of tumor cells, where CD24 plays a crucial role in tumor progression and metastases formation. CD24 aptamers could be used for the detection of cancer cells in earlier states and to evaluate cancer development. Based on former findings using antibodies, targeting of CD24 with aptamers might also cause apoptosis in cancer cells, representing an alternative avoiding the risks going along with the use of antibodies, for example, acute life-threatening allergic reactions [34]. In that case, aptamers also could be used in combination with common anticancer therapeutics.
Footnotes
Acknowledgments
We are thankful to Udo Schumacher, Tobias Lange, and Daniel Wicklein (Institute of Anatomy and Experimental Morphology, University Medical Center Hamburg-Eppendorf) for providing us with all cell lines used in this project. We also thank Kristina Szameit for the scientific support.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.