
Abstract
Recently, our group reported that a small interfering RNA (siRNA) targeting coagulation factor XII (siF12) leads to an unexpected prothrombotic response in a mouse model where venous thrombosis follows inhibition of endogenous anticoagulants. In this study, we aimed to clarify this unexpected response by evaluating the effects of this siF12 (here, siF12-A) on plasma coagulation through thrombin generation (TG). Besides a routine negative control siRNA (siNEG), we included extra siRNA controls: one siRNA similar to siF12-A except for positions 9–11 of the siRNA that are replaced with its complementary base pairs (siF12-AC9/11), and a second siRNA against F12 (siF12-B). Three days after injection, a significant increase in TG peak height was observed solely for animals injected with siF12-A and siF12-AC9/11, which is considered prothrombotic. As this change in coagulation was unrelated to FXII we conclude that it was off-target. For siRNA studies we now recommend to include mismatch siRNA controls, such as the C9/11 mismatch control used in this study, and to consider plasma coagulation in off-target analysis.
Introduction
Inhibition of the liver-produced coagulation factor XII (FXII) has been proposed as a novel therapy in the prevention and treatment of venous thrombosis, a disease in which unwanted blood coagulation in the venous vasculature leads to the formation of a thrombus (www.who.int). In contrast to current “antithrombotic” therapies, FXII inhibition is anticipated not to coincide with bleeding as an undesired side effect, which is common in all current anticoagulant therapies [1–3]. In line with this observation, our group aimed to evaluate the role of FXII in a mouse model for venous thrombosis, which is based on inhibition of the natural (endogenous) anticoagulants antithrombin and protein C. In this model for venous thrombosis, wild-type (WT) mice developed a venous thrombotic coagulopathy, most notably in the veins of the head, without any additional interventions [4].
To test the role of FXII in our mouse model for venous thrombosis, we used a synthetic small interfering RNA (siRNA) targeting exon 3 of the FXII-producing mRNA (gene: F12). This siRNA against F12 (siF12) was selected based on its efficiency for inhibiting F12 in vitro, using primary hepatocytes from female C57BL/6J mice. Within our siRNA experiments, we controlled siRNA delivery in parallel with a nontargeting siRNA (siNEG). This commercially available siNEG has minimal sequence similarity to known genes, is validated for use in human, mouse, and rat cells, and is functionally proven to have minimal effects on cell proliferation and viability (https://thermofisher.com/order/catalog/product/4390844). Including siNEG in our experiments should allow suitable control conditions for the procedure, delivery vehicles, chemistry, and activation of the RNA-induced silencing complex (RISC) machinery. The siRNAs were delivered in vivo to the liver parenchymal cells using invivofectamine® [4,5].
As expected based on in vitro studies, siF12 effectively silenced liver F12 transcript and lowered FXII plasma levels without any detectable changes in mouse health or liver function, while siNEG did not alter F12 transcript or FXII plasma protein levels (see retracted study [6,7]). Conversely, we found that siRNA-mediated lowering of FXII using siF12 accelerated and exacerbated the venous thrombotic phenotype of mice, when compared with the siNEG treated mice. This was surprising and in strong contrast with previous observations where inhibiting FXII resulted in thromboprotection [8–10]. In the current study, we dug deeper into our remarkable finding and now report that the siF12 used in our previous study [6,7] induced a sequence-related procoagulant change in mouse plasma, which appeared a false-positive FXII-independent off-target effect. We conclude that siNEG as a control is insufficient for (in vivo) siRNA studies and recommend to include mismatch controls, such as the C9/11 mismatch control used in our study. Furthermore, we suggest to consider plasma coagulation in off-target analysis.
Methods
Mouse experiments
WT C57BL/6J female mice (18–20 g) were purchased from Charles River (Maastricht, The Netherlands). F12-deficient mice [2,3] were backcrossed to C57Black/6J and WT controls were bred at the University Medical Center Hamburg. Sequences of the siRNAs that were employed in this study (Ambion Silencer select; Thermo Scientific, Waltham, MA) are in Table 1, except for siNEG (catalog number 4390844) for which the sequence is not provided by the manufacturer. For in vivo use, siRNAs were complexed with Invivofectamine 3.0 (Thermo Scientific), according to the manufacturer's instructions. siRNAs were injected intravenously (tail vein) at a dose of 1.2 mg siRNA/kg body weight. Pilot studies showed that to achieve similar knockdown of F12 when compared to siF12-A, siF12-B needed to be mixed with siNEG at a ratio of 1:2 (siF12-B:siNEG). Mice were sacrificed 3 days after siRNA injection, and citrated blood and liver were collected as described [11,12]. Of note, all siRNA experiments were performed at the animal facilities of the Leiden University Medical Center, while blood and tissue collection from F12-deficient mice and WT controls was performed at the University Medical Center Hamburg. Upon sacrifice, plasma and frozen tissues were shipped to the Leiden University Medical Center for further analysis. All experimental procedures were approved by the institutional animal welfare committees.
Small Interfering RNA Sequences of siF12-A, siF12-AC9/11, and siF12-B
For the sense-strand, two thymines are added. For the antisense strand, the additional two base pairs are complementary with the target mRNA. Nucleotides 9–11, the bases swapped for the C9/11 variant, are indicated in bold. Of note, the sequence of the routine control siNEG is not provided by the manufacturer.
Capital letters indicate complementary base pairs of the siRNA, while lower case letters indicate the 3′ overhang of the siRNA.
siRNA, small interfering RNA.
Quantitative polymerase chain reaction
Hepatic transcript analysis using quantitative polymerase chain reaction measurements were performed as described in [11,12], with Actb as a house keeping gene. F12 detection: Forward primer: AATCCGTGCCTTAATGGGGG, reverse primer: TCATAGCAGGTCGCCCAAAG. Primers for transcripts of F2, F10, F11, Klkb1, Cyp4v3, Fga, Fgg, Serpine1, and Serpinc1: see Safdar et al. [12].
Plasma analysis
Plasma thrombin generation (TG) was performed with the calibrated automated thrombography method of [13], using a Fluoroskan Ascent Fluorometer (Thermo Scientific) equipped with a dispenser (Thrombinoscope BV, Maastricht, The Netherlands). Mouse plasma samples (20 μL) were supplemented with 30 μL dilution buffer (20 mM HEPES, 150 mM NaCl, 0.1% PEG8000, pH 7.5), containing 4 μM phospholipids and activator. Activators were tissue factor (TF, 1 pM; Thrombinoscope BV) or ellagic acid (EA, 20 μg/mL; Sigma-Aldrich, Saint Louis, MO). After 10 min of incubation, TG was started with 10 μL FluCa (Thrombinoscope BV). TG was calculated using the Thrombinoscope software package (version 5.0.0.742).
Fibrinogen and serum amyloid A (SAA) plasma levels were determined using commercial ELISA kits (Affinity Biologicals, Ancaster, Canada and R&D systems, Minneapolis, MN, respectively). Plasma levels of the liver enzymes aspartate-amino transferase (ASAT), alkaline phosphatase (ALP), alanine transaminase (ALAT), gamma-glutamyl transferase (GGT), and lactate dehydrogenase (LDH) were determined in 1:3 diluted mouse plasma by routine clinical chemistry analysis at the Department of Clinical Chemistry (Leiden University Medical Center). Antithrombin plasma levels were determined using the Antithrombin III murine ELISA kit (Stago, Leiden, The Netherlands). Using standard western blotting techniques, plasma FXII was detected with the 3F7 antibody [14], plasma prekallikrein (PPK) with the SAPK-PK antibody (Affinity Biologicals), and plasma coagulation factor XI (FXI) with the 14E11 antibody [15].
Statistical analysis
Statistical analysis was performed in Graphpad Instat (GraphPad Software, La Jolla, CA). Data are presented with median and range. Statistical differences were assessed using the Mann–Whitney U test or Kruskall–Wallis multiple comparisons test. A P-value <0.05 was considered statistically significant.
Results and Discussion
In a mouse model for venous thrombosis, which is based on the inhibition of natural (endogenous) anticoagulants antithrombin and protein C, inhibiting the hepatic production of FXII using an siRNA (siF12) resulted in an unexpected and counterintuitive acceleration and exacerbation of venous thrombosis, as compared to siNEG treated mice [6,7]. In contrast, genomic deletion of FXII (F12-deficient mice) did not evoke an acceleration or worsening of venous thrombosis in this mouse model [16]. These results indicated that the reported prothrombotic effect of low FXII could be limited to the siRNA approach.
To clarify why siRNA-mediated F12 silencing, in contrast to full F12 genomic deletion, accelerated and exacerbated venous thrombosis, WT female C57BL/6J mice were treated with siF12 (in a setting without a thrombotic challenge). Three days after siF12 injection, F12 transcript levels were lowered to 11.4% (range: 7.8; 15.8) when compared with siNEG treated mice (siNEG: 100%, P < 0.001, Fig. 1A). At this timepoint, other liver transcript levels for a number of coagulation and acute-responder genes were normal and comparable (Supplementary Fig. S1A). In addition, liver-produced plasma protein levels of FXII-targets PPK and FXI, inflammation-related proteins fibrinogen, SAA, and the liver enzymes ASAT, ALP, ALAT, GGT, and LDH were considered normal or below the detection limit, and comparable between all groups independent of siRNA treatment (Supplementary Fig. S1B–G). These results imply that siNEG and siF12 treatment did not evoke (aspecific) changes in the plasma or liver parameters analyzed.
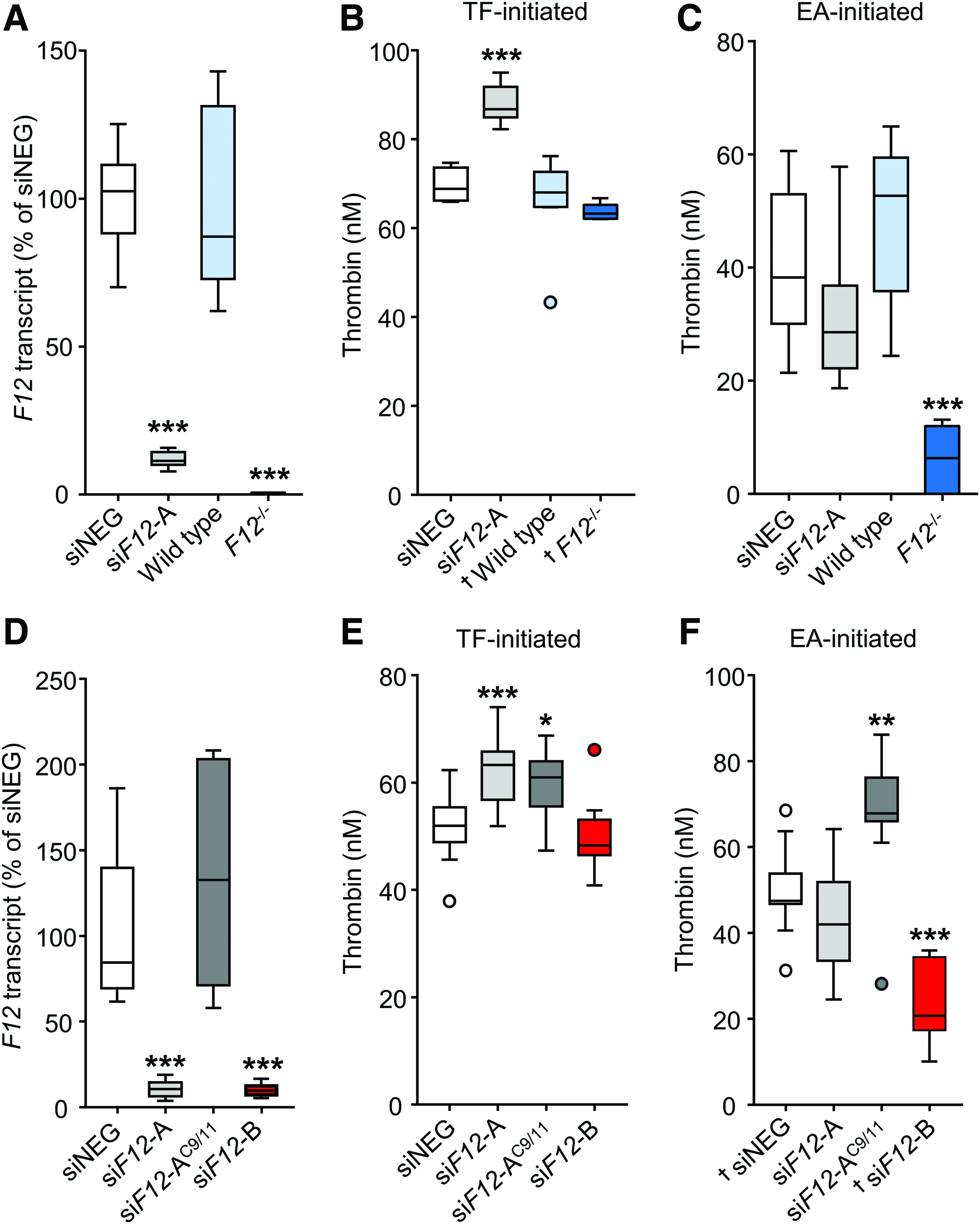
siF12-A treatment coincides with a FXII-independent procoagulant response in mice.
To evaluate coagulation of siRNA treated mice in more detail, mouse plasmas were subjected to a TG assay. TG analysis allows global assessment of the balance between pro- and anticoagulation upon stimulation of plasma coagulation by endogenous or exogenous initiators of coagulation (TF and EA, respectively), and it is a well-established analytical tool within thrombosis and hemostasis research [17]. In short, in addition to plasma clotting time, a parameter that can be measured in all traditional coagulation tests, TG can be used to determine the highest thrombin concentration that can be generated (peak height), velocity of TG (time to peak), and total amount of thrombin that can be generated (area under the curve). These parameters are dependent on the potency of plasma pro- and anticoagulants [13,18]. Using TG human (or murine) plasma can be tested for clinical conditions associated with thrombosis [19,20]. For TG measurements, plasma from WT (normal F12 levels) and F12-deficient mice [F12−/−, no detectable F12 transcript (Fig. 1A) and FXII plasma protein] were included as additional controls.
Three days after siRNA injection, when initiating TG in plasma of siF12-injected mice using TF, TG peak height demonstrated an unexpected significant increase when compared with siNEG treated mice [siNEG; 68.9 nM (65.9; 74.7) and siF12: 86.8 nM (82.3; 95.0), P < 0.001, Fig. 1B and Supplementary Fig. S2A], which can be considered as a prothrombotic change. This observation was reproduced in multiple independent experiments with an increase of TF-induced TG peak height of ∼20%, although TF-induced TG is reportedly independent on FXII activity ([21] and own observations). EA-induced TG of plasma from siF12 mice was not altered [siNEG; 38.2 nM (21.4; 60.0) and siF12: 28.6 nM (18.7; 57.9), P = 0.055, Fig. 1C and Supplementary Fig. S2B]. This was unexpected, since EA activates FXII that leads to thrombin formation in plasma, making TG strongly dependent on FXII activity ([8,22,23] and own observations).
To exclude that the siF12-related impact on TG is the result of siF12 effects other than FXII-lowering, that is, off-target FXII-independent effects of siF12, two additional control siRNAs were designed: One siRNA was similar to the original siF12 (from now on referred to as siF12-A) except for nucleotides 9–11, which were replaced with their complementary base pairs: siF12-AC9/11. Since the seed sequence of siF12-A is unaltered in the siF12-AC9/11 mismatch control, false-positive siF12-A off-target effects will likely maintain their activity, whereas true positive siF12-A on-target effects will lose impact [24]. In addition, a second siRNA targeting F12 was used (siF12-B), which targets exon 9 of the F12 mRNA. siF12-B showed similar efficiency for inhibiting F12 during our in vitro selection using mouse primary hepatocytes (data not shown). In vivo, 3 days after injection, siF12-B showed to inhibit F12 even more potent than siF12-A (Supplementary Fig. S3). Of note, siF12-A, siF12-AC9/11, and siF12-B were designed not to target other mouse transcripts, which was confirmed by BLAST.
WT mice were treated with siNEG, siF12-A, siF12-AC9/11, or siF12-B (1:2 diluted with siNEG to match siF12-A inhibition of F12). Three days after siRNA injection, liver transcript analysis confirmed that siF12-A and (diluted) siF12-B treated mice had equally strong reduced F12 levels, while for control siF12-AC9/11 injected mice F12 was not affected [compared to siNEG, siF12-A: 10.8% (3.9; 19.2), siF12-AC9/11: 132.8% (58.1; 208.4), siF12-B: 10.0% (5.5; 16.6), P < 0.001, Fig. 1D]. Remarkably, plasma from both siF12-A and siF12-AC9/11 injected mice showed a strong procoagulant shift in TG peak height upon TF activation [siNEG: 52.0 nM (37.9; 62.4), siF12-A: 63.3 nM (51.1; 74.0), siF12-AC9/11: 61.0 nM (47.3; 68.8), P < 0.05, Fig. 1E and Supplementary Fig. S2C]. Plasma from siF12-AC9/11 injected mice responded aberrant when TG was initiated by EA, while despite low levels of FXII plasma from siF12-A treated mice was again not different from siNEG-injected mice in TG peak height [siNEG: 47.5 nM (31.3; 68.6), siF12-A: 42.0 nM (24.5; 64.1), siF12-AC9/11: 67.8 nM (28.2; 86.2), P < 0.01, Fig. 1E and Supplementary Fig. S2C]. In contrast to siF12-A but similar to FXII-deficient plasma, siF12-B treatment did not influence the peak height in TF-induced plasma TG [siNEG: 52.0 nM (37.9; 62.4), siF12-B: 48.3 nM (40.9; 66.1), P = 0.22], while EA-induced TG was strongly decreased in siF12-B plasma [siNEG: 47.5 nM (31.3; 68.6), siF12-B: 20.7 nM (10.1; 35.9), P < 0.001, Fig. 1F and Supplementary Fig. S2D]. These results indicate that the observed prothrombotic shift in plasma TG upon siF12-A treatment cannot be attributed to FXII and appears a false-positive FXII-independent off-target effect.
A prolonged “time to tail” in the curves of TF-induced TG is observed for siF12-A and siF12-AC9/11, which suggests that the siRNAs affect the plasma's ability to inhibit thrombin (Supplementary Fig. S2B, D). In line with this observation, plasma levels of the endogenous thrombin-inhibitor antithrombin were decreased for siF12-A and siF12-AC9/11 treated mice [compared to siNEG, siF12-A: 70.7% (21.7; 85.4), siF12-AC9/11: 74.4% (61.1; 85.1), siF12-B: 101.9% (87.2; 118.3), P < 0.001], which could at least partly explain the effects on TG. Of note, this would be without an effect on antithrombin mRNA (gene: Serpinc1, Supplementary Fig. S4), which implies consumption of antithrombin in plasma.
The exact mechanism via which the off-target effect of siF12-A and siF12-AC9/11 operates is currently unknown. Since the seed sequence was unaltered between siF12-A and siF12-AC9/11, the seed sequence is a likely candidate responsible for the procoagulant (off-target) effect of the siRNAs [25–27]. Bioinformatical analysis (using BLAST software) for the sense and antisense strand of the siRNA did not yield complementary candidates for the mouse (or human) transcriptome (besides the mouse F12 gene). Homology of the seed sequence to the mouse transcriptome might explain the off-target effect. However, the seven-base seed sequence is too short for BLAST analysis. When using BLAST for the seven-base seed sequence would be feasible, this analysis would very likely produce a multitude of candidate (off-) targets precluding adequate interpretation of the in silico analysis.
An alternative approach to investigate whether the seed sequence is responsible for the procoagulant off-target is to develop a mismatch control with an altered seed sequence. However, we argue that such an extra siRNA control should always be used in addition to a mismatch control (eg, C9/11), since otherwise the seed sequence-specific off-target effect would not be identified and stay unnoticed. Despite that our study is not conclusive as no seed sequence disruption studies were included, we suggest to avoid the seed sequence of siF12-A and siF12-AC9/11 that is, UUGUGGA (antisense) in RNAi studies. Generally, studies for seed sequence off-target regulation should be performed routinely as part of identifying specific, safe siRNA molecules. This recommendation holds true for studies in mice, the species in which the siF12-A and siF12-AC9/11 off-target effect was detected, but also for studies in other species that is, humans. In addition, future studies have to decipher whether siF12-A and siF12-AC9/11 are unique in provoking a procoagulant response or whether this response is more global and applies to multiple siRNAs. Such studies may also yield insight into known and unknown aspects of blood coagulation regulation.
We conclude that treatment of mice with the siRNA targeting exon 3 of F12 ([6,7], here siF12-A) induced unexpected off-target effects unrelated to FXII inhibition, which became evident upon including plasma TG analysis. Whether this siRNA-related effect on coagulation is confined to the siRNAs studied here or is more common is currently unknown. However, based on our results we recommend to consider plasma coagulation as a potential pathway for siRNA off-target effects. In addition, for studies using siRNA we advise to include a mismatch control, like the C9/11 mismatch siRNA used in our study, which provides the ability to distinguish between true and false positives. Along these lines, when studying FXII using siF12-B, a control such as siF12-BC9/11 would be appropriate. While siNEG-type siRNAs control for the experimental procedure, delivery vehicles, chemistry, and activation of the RISC machinery, it appears to have insufficient ability to completely distinguish between true and false positives.
Footnotes
Acknowledgments
The authors would like to thank Dr. Thomas Renné (Institute for Clinical Chemistry, University Hospital Eppendorf, Hamburg, Germany) for providing materials and mice, Dr. David Gailani (Department of Medicine, Vanderbilt University Medical Center, Nashville, TN) for providing the 14E11 antibody, Dr. René van Oerle (Department of Internal Medicine and Biochemistry, Maastricht University, Maastricht, the Netherlands) for technical assistance, and Dr. Wim Reidt (Thermo Scientific) for help in siRNA design.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.