
Abstract
Antisense oligonucleotides (ASOs) hold promise for therapeutic splice switching correction for genetic diseases, in particular for Duchenne muscular dystrophy (DMD), for which ASO-exon skipping represents one of the most advanced therapeutic strategies. We have previously reported the therapeutic potential of tricyclo-DNA (tcDNA) in mouse models of DMD, highlighting the unique pharmaceutical properties and unprecedented uptake in many tissues after systemic delivery, including the heart and central nervous system. TcDNA-ASOs demonstrate an encouraging safety profile and no particular class-related toxicity, however, when administered in high doses for several months, mild renal toxicity is observed secondary to predictable phosphorothioate (PS)-ASO accumulation in kidneys. In this study, we investigate the influence of the relative content of PS linkages in tcDNA-ASOs on exon skipping efficacy. Mdx mice were injected intravenously once weekly for 4 weeks with tcDNA carrying various amounts of PS linkages (0%, 25%, 33%, 50%, 67%, 83%, and 100%). The results indicate that levels of exon-23 skipping and dystrophin rescue increase with the number of PS linkages in most skeletal muscles except in the heart. As expected, plasma coagulation times are shortened with decreasing PS content, and tcDNA-protein binding in serum directly correlates with the number of PS linkages on the tcDNA backbone. Altogether, these data contribute in establishing the appropriate sulfur content within the tcDNA backbone for maximal efficacy and minimal toxicity of the oligonucleotide.
Introduction
Antisense strategies are based on the use of antisense oligonucleotides (ASOs) to modify mRNA by increasing or decreasing gene expression or via splicing modulation [1]. ASO-based therapeutics are currently one of the most promising therapeutic solutions for the treatment of many yet-uncured genetic diseases, in particular Duchene muscular dystrophy (DMD) for which ASO-exon skipping represents one of the most advanced therapeutic strategies [2–4].
ASOs were originally unmodified DNA sequences but have rapidly evolved to prevent degradation within cells and bloodstream [5]. Phosphorothioate (PS) backbones were the first and remain one of the most largely used chemical modifications to protect ASOs from nuclease activity and increase their stability [6]. Typical PS differs from phosphodiester (PO) bonds by the replacement of one of the nonbridging-phosphate O-atoms with an S-atom, which confers higher stability and increased cellular uptake [7]. PS modifications have demonstrated improved efficacy due to an increased bioavailability compared with their PO counterparts [8] and most ASOs currently under clinical programs include PS bonds [9]. Nevertheless, despite their pharmacokinetic (PK) advantage, PS-modified molecules are known to cause toxicity or undesirable effects mainly due to their capacity to bind plasma proteins [10]. Acute reactions/effects of PS backbones may include immune cell activation [11], complement activation (particularly reported in monkey studies) [12], or prolongation of clotting times [10,13] that is known to be transient and normalize as oligonucleotides are cleared from the bloodstream. Notably, it has been shown that low-level, but sustained, complement activation may lead to depletion of complement and damage to the vascular and renal system [14,15]. In addition, the increased protein binding of PS-oligonucleotides prevents their rapid excretion in the urine, as they form large complexes that impair their filtration by the glomerulus. Following repeated-dose systemic administration, PS-ASOs typically target organs such as the kidney and liver where they accumulate and may cause cellular toxicity.
We have previously demonstrated the therapeutic potential of a novel class of ASO for DMD, the tricyclo-DNA (tcDNA) that displays unique pharmacological properties and unprecedented uptake in many tissues after systemic administration in mouse models of DMD [16,17]. Systemic delivery of full PS-tcDNA (15-mer or 13-mer) allows restoration of dystrophin in skeletal muscles and to a lower extent in the brain, leading to muscle function improvement and correction of behavioral features linked to emotional/cognitive deficiency. Although PS-tcDNA-ASOs present an overall encouraging safety profile, high-dose treatment with PS-tcDNA over several months led to slightly increased serum creatinine values in treated mice compared with wild-type (WT) and mdx controls. In addition, histopathological findings revealed limited-to-minimal glomerular changes in kidney and moderate liver inflammation in treated mice, typically induced by PS-ASO accumulation within these organs [17]. While this level of toxicity remains very mild and induced with only very high-dose regimens, which would never be used in humans, we aimed to investigate the possibility of reducing PS content in tcDNA-ASO. This is of particular interest in the case of tcDNAs, which are stable in their full PO version as opposed to other PO-based ASOs, which require PS bonds for stability [18,19].
In this study, we explore the potential of minimizing PS-associated toxicity and aim to evaluate whether the number of PS linkages can be reduced without compromising tcDNA exon skipping efficacy. TcDNA compounds targeting exon 23 of the mouse dmd gene and carrying different amounts of PS linkages (0%, 25%, 33%, 50%, 67%, 83%, and 100% PS) were designed and their serum protein-binding and exon skipping properties were investigated in vitro and in vivo. We first show the correlation between serum protein binding and the amount of PS bonds within the tcDNA-ASO as well as the reduction of coagulation times with decreased number of PS bonds. We then evaluate the antisense activity of these compounds administered intravenously (IV) during 4 weeks in the mdx mouse model. We show that tissue biodistribution and exon skipping efficacy are higher with increased number of PS in all tested tissues except in the heart, which appears to be less sensitive to PS content. Interestingly, tcDNA-ASOs containing from 50% to 67% of PS linkages induce similar levels of exon-23 skipping compared with the full PS counterpart, suggesting the possibility of reducing PS content without significantly affecting the efficacy of tcDNA.
Materials and Methods
ASO and animal experiments
Animal procedures were performed in accordance with the national and European legislation, approved by the French government (Ministère de l'Enseignement Supérieur et de la Recherche, Autorisation APAFiS #6518). Mdx (C57BL/10ScSc-Dmdmdx/J) mice were bred in our animal facility at the Plateforme 2Care, UFR des Sciences de la santé, Université de Versailles-Saint Quentin and were maintained in a standard 12-h light/dark cycle with free access to food and water. Mice were weaned at weeks 4–5 postnatal and 2–5 individuals were housed per cage.
All tcDNA-ASOs targeting the donor splice site of exon 23 of the mouse dystrophin pre-mRNA, used in this study, are detailed in Table 1 and were synthesized by SYNTHENA (Bern, Switzerland). The distribution of PS linkages along with the tcDNA oligonucleotides appears to have little impact on exon skipping or biodistribution, as shown in Supplementary Fig. S1. For this work, we therefore randomly picked a 3′ design, that is, where the number of PS linkages is progressively reduced from the 5′-end of the tcDNA sequences (Table 1).
tcDNA Oligonucleotide Sequences
Corresponds to PS link.
tcDNA, tricyclo-DNA; PS, phosphorothioate; PO, phosphodiester.
Eight groups of four 6–8-week-old mdx mice were injected IV in the retro-orbital sinus, under general anesthesia using 1.5%–2% isoflurane, once a week with the corresponding tcDNA for a period of 4 weeks (Fig. 4c). An age-matched mdx group receiving an equivalent volume of sterile saline was included as control. One hour after the first injection, blood samples were collected from all mice to measure complement C3. Additional blood samples were collected 1 week after the end of the treatment. Animals were euthanized 2 weeks after the end of the 4-week treatment and muscles and tissues were harvested and snap-frozen in liquid nitrogen-cooled isopentane and stored at −80°C before further analysis. For PK studies, additional mdx mice were injected IV with the different compounds (single injection) and serum was collected at different time points, as indicated in Fig. 4a (n = 3 mice per time point). Sample sizes and n values are indicated in each figure legend. Investigators were blinded for RNA and protein analysis.
Serum analysis
Analyses of serum creatine kinase (CK), alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), bilirubin, creatinine, urea, and albumin levels were performed by the pathology laboratory at Mary Lyon Centre, Medical Research Council, Harwell, Oxfordshire, United Kingdom. Human serum from healthy volunteers was obtained from the French Blood Donors Organization (Etablissement Français du Sang). Complement activation in mouse serum samples was measured by the MicroVue PS-C3 converter and SC5b-9 Plus kits (Quidel Co., San Diego, CA). For in vitro complement activation studies, tcDNA was incubated with mouse or human serum at 37°C for 45 min. Determination of complement activation was evaluated using the mouse C3a ELISA kit (TECO Medical, Switzerland) and human C3aPlus kit (Quidel Co.). Zymosan, 5mg/ml (Complement Technology, Inc., Texas), was used as positive control.
Coagulation assays
Mouse citrated plasma samples were incubated in vitro with 2 mg/mL of tcDNA for 20 min at 37°, and then, prothrombin time (PT) and activated partial thromboplastin time (aPTT) assays were performed on a semiautomated START max coagulometer (Stago) following the manufacturer's instructions.
Cell transfection and gymnosis
Mdx mouse muscle cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 20% fetal bovine serum and 1% penicillin/streptomycin (100 U/mL). Twenty-four hours before gymnosis, mdx cells are plated in 12-well plates (8 × 104 cells per well). Cells were then incubated for 72 h in a differentiation medium (DMEM, 4% horse serum, 1% penicillin/streptomycin) with 30 μM of tcDNA-ASOs and harvested using the TRIzol reagent to isolate total RNA according to the manufacturer's instructions (Thermo Fisher Scientific).
RNA analysis
Total RNA was isolated from muscle cells or intervening muscle sections collected during cryosection using the TRIzol reagent according to the manufacturer's instructions (Thermo Fisher Scientific). Aliquots of 500 ng of total RNA were used for reverse transcription polymerase chain reaction (RT-PCR) analysis using the Access RT-PCR System (Promega) in a 50 μL reaction using the external primers Ex 20Fo (5′- CAGAATTCTGCCAATTGCTGAG-3′) and Ex 26Ro (5′-TTCTTCAGCTTGTGTCATCC- 3′). The cDNA synthesis was carried out at 45°C for 45 min, directly followed by the primary PCR of 30 cycles of 95°C (30 s), 55°C (1 min), and 72°C (2 min). Two microliters of these reactions was then reamplified in nested PCRs by 22 cycles of 95°C (30 s), 55°C (1 min), and 72°C (2 min) using the internal primers Ex 20Fi (5′- CCCAGTCTACCACCCTATCAGAGC-3′) and Ex 26Ri (5′- CCTGCCTTTAAGGCTTCCTT-3′). PCR products were analyzed on 2% agarose gels. Exon 23 skipping was also measured by TaqMan quantitative RT-PCR as previously described, using TaqMan assays that were designed against the exon 4–5 or exon 22–24 templates using the Custom Assay Design Tool (Life Technologies) (Assay Ex4–5: Forward: 5′- GGCACTGCGGGTCTTACA-3′; Reverse: 5′- CATCCACTATGTCAGTGCTTCCTAT 3′; Probe: 5′- TTCACTAAATCAACATTATTTTTC -3′ and assay Ex22–24: Forward: 5′- CTGAATATGAAATAATGGAGGAGAGACTCG -3′; reverse: 5′- CTTCAGCCATCCATTTCTGTAAGGT-3′; Probe: 5′- ATGTGATTCTGTAATTTCC-3′). An inventoried 18S assay was utilized as an endogenous control (Life Technologies). Fifty nanograms of cDNA was used as input per reaction and all assays were carried out in triplicate. Assays were performed under fast cycling conditions on a BioRad CFX96 Touch Real-Time PCR Detection System, and all data were analyzed using the comparative Ct method. For a given sample, the delta-Ct values of exon 4–5 and exon 22–24 assays were used to calculate a relative abundance of total dystrophin and exon 23-skipped dystrophin mRNA, respectively. Exon 23 skipping was then expressed as a percentage against total dystrophin, as indicated by the exon 4–5 expression level.
Western blot
Protein extracts were obtained from pooled muscle sections treated with RIPA lysis and extraction buffer (Thermo Fisher Scientific) complemented with sodium dodecyl sulphate (SDS) powder (5% final) (Bio-Rad, France), and the total protein concentration was determined with the BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were denatured at 100°C for 3 min and 50 μg of protein was loaded onto NuPAGE 3%–8% Tris-acetate protein gels (Invitrogen), following the manufacturer's instructions. Dystrophin protein was detected by probing the membrane with the NCL-DYS1 primary monoclonal antibody (NCL-DYS1; Novocastra, Newcastle, United Kingdom) and vinculin was detected as internal control with the hVin-1 primary antibody (Sigma), followed by incubation with a goat anti-mouse secondary antibody (IRDye 800CW goat anti-mouse IgG, Li-Cor, Germany). Bands were visualized using the Odyssey CLx system (Li-Cor, Germany). The quantification was done using standard curve with 0%, 5%, 10%, and 20% of corresponding WT tissues and normalized to internal control (vinculin).
tcDNA quantification by fluorescent hybridization assay
Tissues were homogenized in proteinase K buffer (100 mmol/L Tris–HCl, pH 8.5, 200 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.2% sodium dodecyl sulfate) containing 2 mg/mL of proteinase K (Invitrogen) (50 mg tissue/mL of buffer), followed by incubation overnight at 55°C in a hybridization oven. After centrifugation at 14,000 rpm (Sorvall ST 8R centrifuge, 75005719 rotor) for 15 min, the supernatant was used in the assay. Quantification of tcDNA was performed using a hybridization assay with a molecular beacon probe, based on the previously published technology [20]. Briefly, 10 μL of tissue lysates or serum was incubated with a 5′ Cy3-DNA complementary probe conjugated with HBQ quencher at 3′ in a black nonbinding 96-well plate (Fischer Scientific). Phosphate buffered saline (PBS) was added to a final volume of 100 μL per well and fluorescence was measured on a spectrophotometer (Ex 544 nm/Em 590 nm using FLUOstar Omega). The amount of tcDNA in tissues was determined using a standard curve built on the measurement of known tcDNA quantities dissolved in the respective tissue lysates of mock-injected animals.
PK analysis of serum concentration
For PK studies, 6–8-week-old mdx mice received a single intravenous injection of full PS-tcDNA (100% PS) or full PO-tcDNA (0% PS) and blood samples were collected as detailed in Fig. 4a (n = 3/time point). PK was analyzed using WinNonlin 8.1 software (Pharsight Corporation, Mountain View, CA). Semilogarithmic plots of tcDNA serum level means versus time indicated biexponential decrease.
Protein binding analysis
Analysis of serum protein binding to the oligonucleotides was performed with the biotinylated compounds immobilized on high-capacity streptavidin agarose beads (Thermo Fisher). Immobilized oligonucleotides (20 μL of high-capacity streptavidin agarose beads/10 μg of oligonucleotide) were incubated with 50 μL of mouse serum for one hour. Beads were collected by centrifugation and washed four times with 1 × PBS. The precipitate was solubilized in the Laemmli sample buffer and proteins were separated by electrophoresis onto a 4%–12% gradient SDS-polyacrylamide gel. After Coomassie staining, the protein content was estimated by Image Master 2D platinum software.
Statistical analysis
Data were analyzed by GraphPad Prism7 software (San Diego) and shown as the mean ± standard error of the mean; “n” refers to the number of mice per group. Comparisons of statistical significance were assessed by nonparametric Mann–Whitney U tests for the comparison of two groups or Kruskal–Wallis for the comparison of three or more groups followed by Dunn's multiple comparison test.
Significant levels were set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
In vitro studies
Serum protein binding pattern of tcDNA-ASOs depending on the PS content
To explore the influence of PS linkages on tcDNA behavior, we designed tcDNA compounds carrying different amounts of PS linkages (from 0% to 100%) using the M23D previously described sequence, targeting the exon 23 of the mouse dmd gene (Table 1). We first investigated the serum protein binding properties of these various tcDNA compounds using 5′-end biotinylated ASO immobilized on streptavidin agarose beads. Interestingly, the pattern of protein binding to PO-tcDNA (0% PS) was identical to the pattern of protein binding to empty beads, indicating that under described experimental conditions, PO-ASOs are practically free of proteins (Fig. 1a). As expected, full PS-tcDNA ASOs bind numerous proteins of different molecular mass in mouse serum, similarly to other PS chemistries such as 2′OMethyl-RNA (2′OMe-PS) (Supplementary Fig. S2). Importantly, comparison of the protein profiles bound to the M23D sequence and PS-tcDNA polyT sequence indicates that protein binding is mainly determined by the presence of PS linkages and not by the primary sequence of the oligonucleotides (Supplementary Fig. S2). Increase in PS content appears to correlate quite linearly with the increase of bound protein quantities (Fig. 1b), although this is less clear between 83% and 100% PS, where the increase in PS content induces only a slight augmentation in protein binding. This suggests that one needs a minimal length of PS part of the compounds to assure sufficient binding affinity for recovering proteins under the experimental conditions.
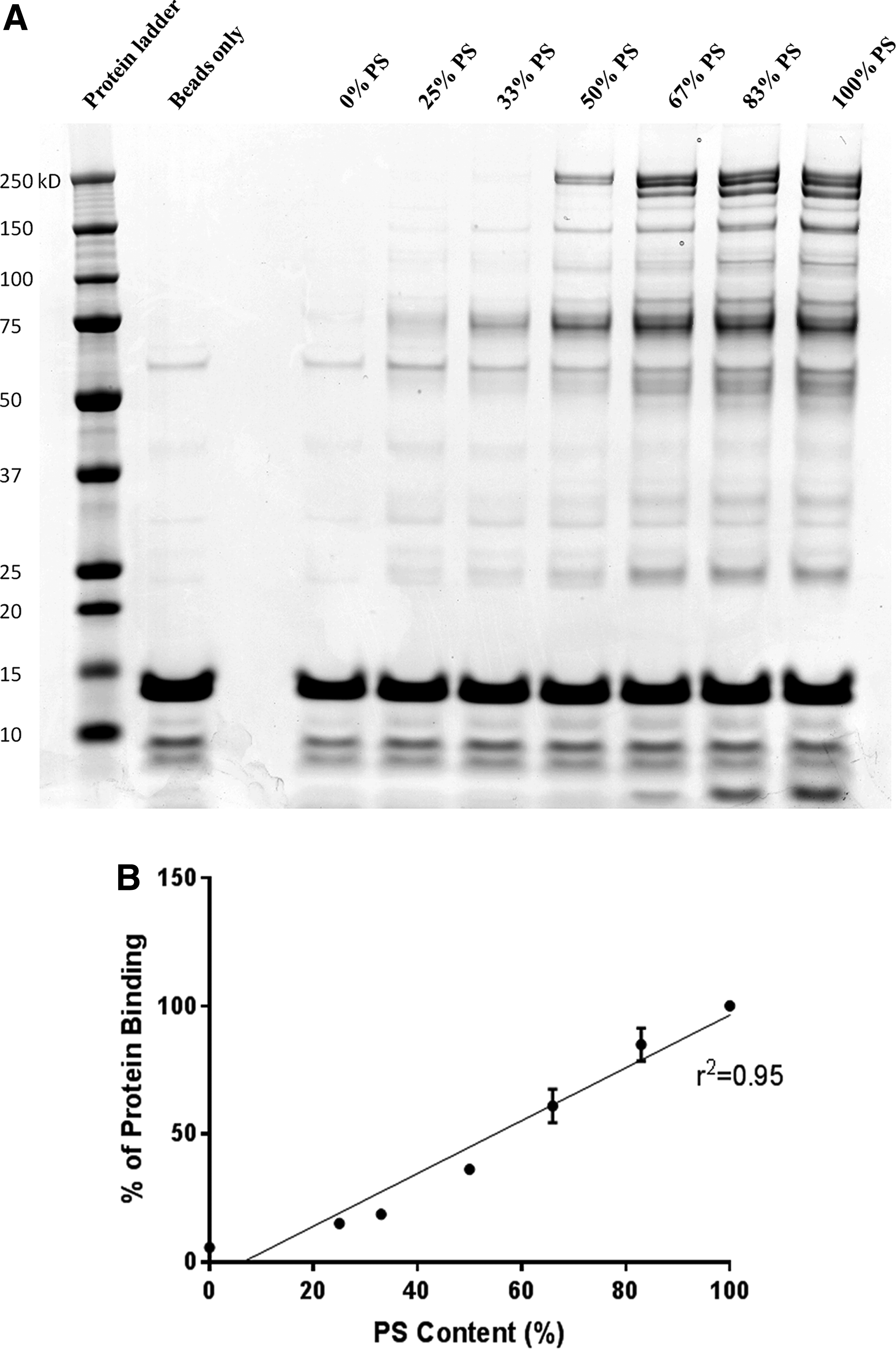
Protein binding to tcDNA oligonucleotides depends on the number of PS linkages. Protein binding was evaluated using biotinylated tcDNAs carrying various contents of PS linkage, from all PO (0%PS) to full PS modifications (100%PS).
Coagulation studies and complement activation
PS-ASOs are known to influence the coagulation cascade and the alternative pathway of the complement system as a protein binding-related effect [12,13]. In this regard, we aimed to evaluate in vitro the intrinsic (aPTT) and extrinsic (PT) coagulation pathways in mouse plasma as well as complement activation in mouse and human serum samples. Appropriate plasma or serum samples were incubated with 2 mg/mL of tcDNAs carrying different amounts of PS modifications (0%, 25%, 33%, 50%, 67%, 83%, and 100% PS). As expected, the results showed prolonged coagulation times with increasing number of PS linkages. Interestingly, the intrinsic pathway (aPTT) is increased by sixfold and already saturated when 50% of the oligonucleotide linkages are PS modified (Fig. 2a). Complement activation analysis revealed no significant decrease in total C3 or increase in C3a split product in vitro in mouse or human serum samples incubated with 100% PS or 0% PS (full PO) (Fig. 2b, c), suggesting that full PS-tcDNA M23D ASO does not activate the complement as opposed to zymosan, which was used as a positive control. These results were confirmed by western blot (WB) (Supplementary Fig. S3).
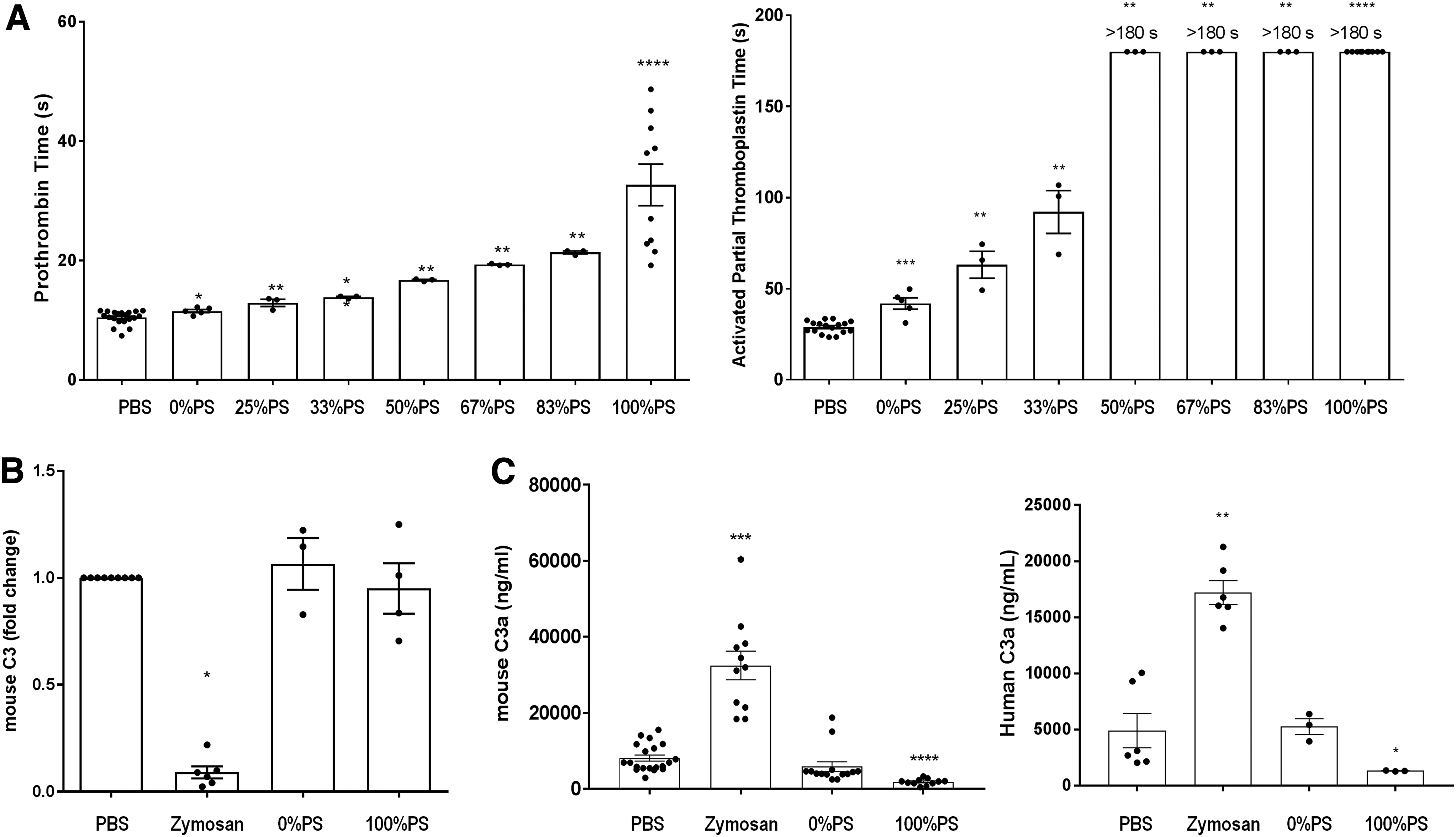
In vitro evaluation of the effect of PS content on clotting times and complement activation.
Gymnotic delivery of ASOs
We next examined the influence of PS modifications on cell and nuclear uptake in cultured mdx mouse muscle cells without transfection agent, a process called gymnosis. TcDNAs carrying varying amounts of PS modifications (0%, 25%, 33%, 50%, 67%, 83%, and 100% PS) were incubated for 72 h with mdx cells, and cells were then collected for quantitative real time PCR (RT-qPCR) analysis. As shown in Fig. 3, exon skipping levels increase with the PS content. TcDNAs carrying 67% and 83% of PS linkages induce similar levels of exon 23 skipping than the compound carrying 100% of PS. On the contrary, the level of exon skipping induced with the compound carrying 25% of PS modification is closer to the level achieved with full PO-tcDNA. Other compounds, 33% and 50% of PS linkages, produce an intermediate level of exon 23 skipping.
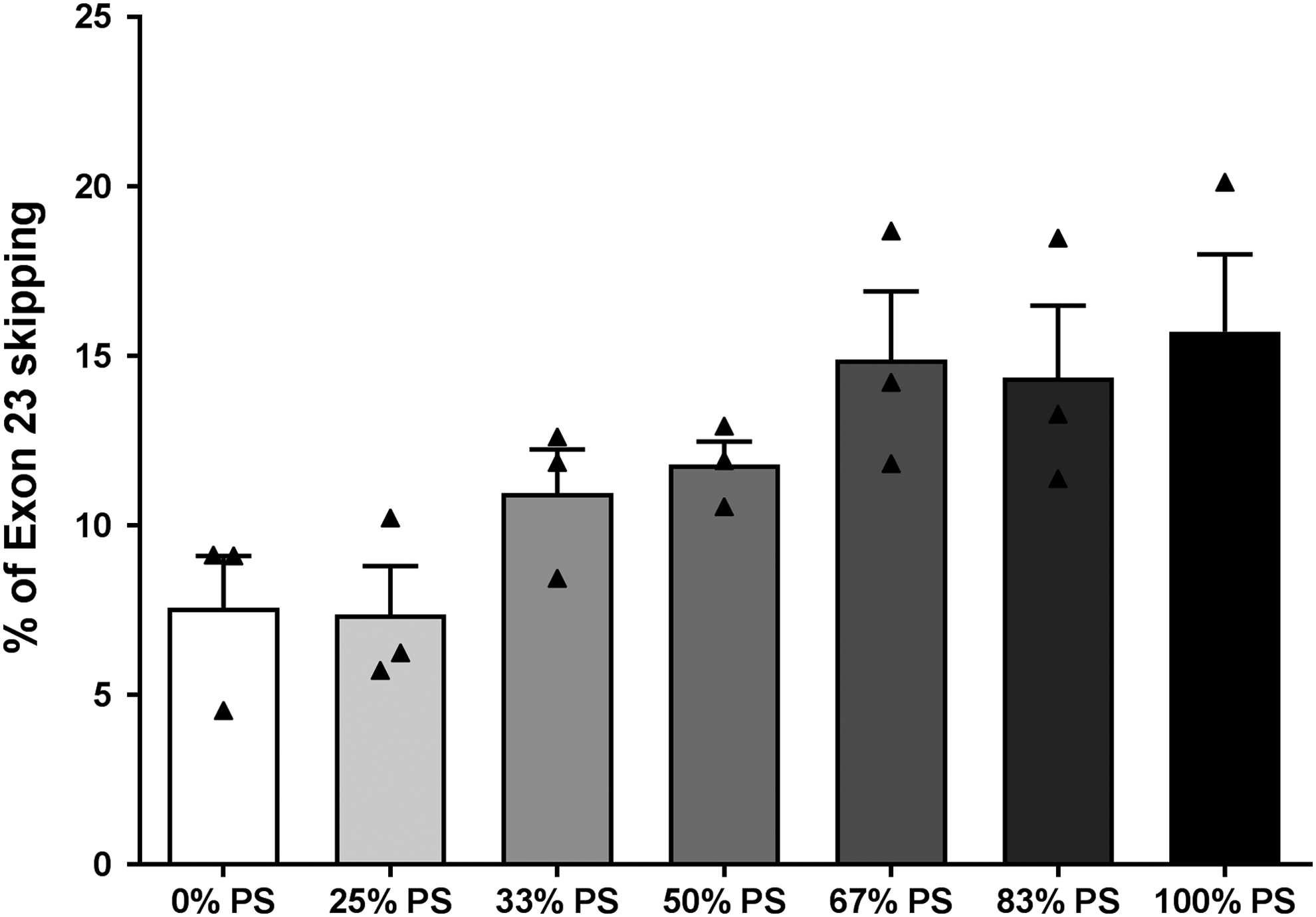
The number of PS modifications correlates with exon 23 skipping efficacy in vitro. Exon 23 skipping level was evaluated by RT-qPCR in mdx mouse cells after passive diffusion (gymnosis) of PS-modified tcDNA carrying 0% PS, 25% PS, 33% PS, 50% PS, 67% PS, 83% PS, and 100% PS. Results are mean ± SEM (n = 3). P > 0.05 between all groups. RT-qPCR, quantitative real time PCR.
In vivo studies
Effect of PS content on tcDNA-ASO PK and biodistribution
To evaluate the contribution of PS content in vivo, the different compounds were injected IV in adult mdx mice, which carry a nonsense mutation in exon 23 of the DMD gene [21]. We first analyzed the PK properties of the full PS-tcDNA (100% PS) and full PO-tcDNA (0% PS) after a single intravenous injection of 50 mg/kg of compounds, as represented in Fig. 4a. PK data on ASOs are fundamental to optimize the dose and regimen appropriate for the future systemic treatment of DMD patients, especially considering that ASO therapy is likely to be chronic. Here, we applied a quantitative and sequence-specific hybridization assay to quantify the levels of tcDNA compounds in the serum of injected mice at different time points. As described for other ASOs [22,23], PK of full PS-tcDNA (100% PS) and full PO-tcDNA (0% PS) was best fitted to biexponential equations and was both characterized by a rapid initial distribution and a long-terminal half-life at very low concentrations (Fig. 4b).

tcDNA quantification in serum and tissues following intravenous injection(s).
We also investigated the biodistribution of the different tcDNA-ASOs in various organs (quadriceps, triceps, diaphragm, heart, kidney, and liver) depending on their PS content. For this purpose, mice were treated IV with 200 mg/kg/week of tcDNA for a period of 4 weeks and their tissues were analyzed 2 weeks after the final injection, as depicted in Fig. 4c. The biodistribution of tcDNA-ASOs appears overall very similar to other ASO chemistry with high levels of tcDNA detected in the liver and kidneys on IV delivery (Fig. 4d; Supplementary Table S1). Quantification of tcDNA-ASOs in various organs demonstrates a clear contribution of PS content in tcDNA accumulation. Increase in PS content up to 67% leads to a small increase of compound delivery to muscles. However, from 83% of PS content, delivery to the quadriceps, triceps, diaphragm, and heart appears substantially improved, reaching a maximum for the full PS compound. Interestingly, amounts of full PS-tcDNAs (100% PS) are significantly higher in all organs than the other ASOs, including the 83% PS-tcDNA (Fig. 4d).
Safety and tolerability
PS ASOs are well known for their immunostimulatory effects [24] and for activating the alternative pathway of complement among others [25]. In this regard, we carried out complement activation studies in mdx mice treated with 200 mg/kg/week of tcDNA carrying different amounts of PS modifications (0% PS, 25%, 33%, 50%, 67%, 83%, and 100% PS). The results show that C3 protein levels, a complement protein with a central role in complement activation, are slightly diminished 1h after injection when 100% PS-tcDNA is injected, although no adverse clinical response to injection had ever been observed. This slight complement activation does not occur when PS linkages are decreased, from 83% to 0% (Fig. 5a).
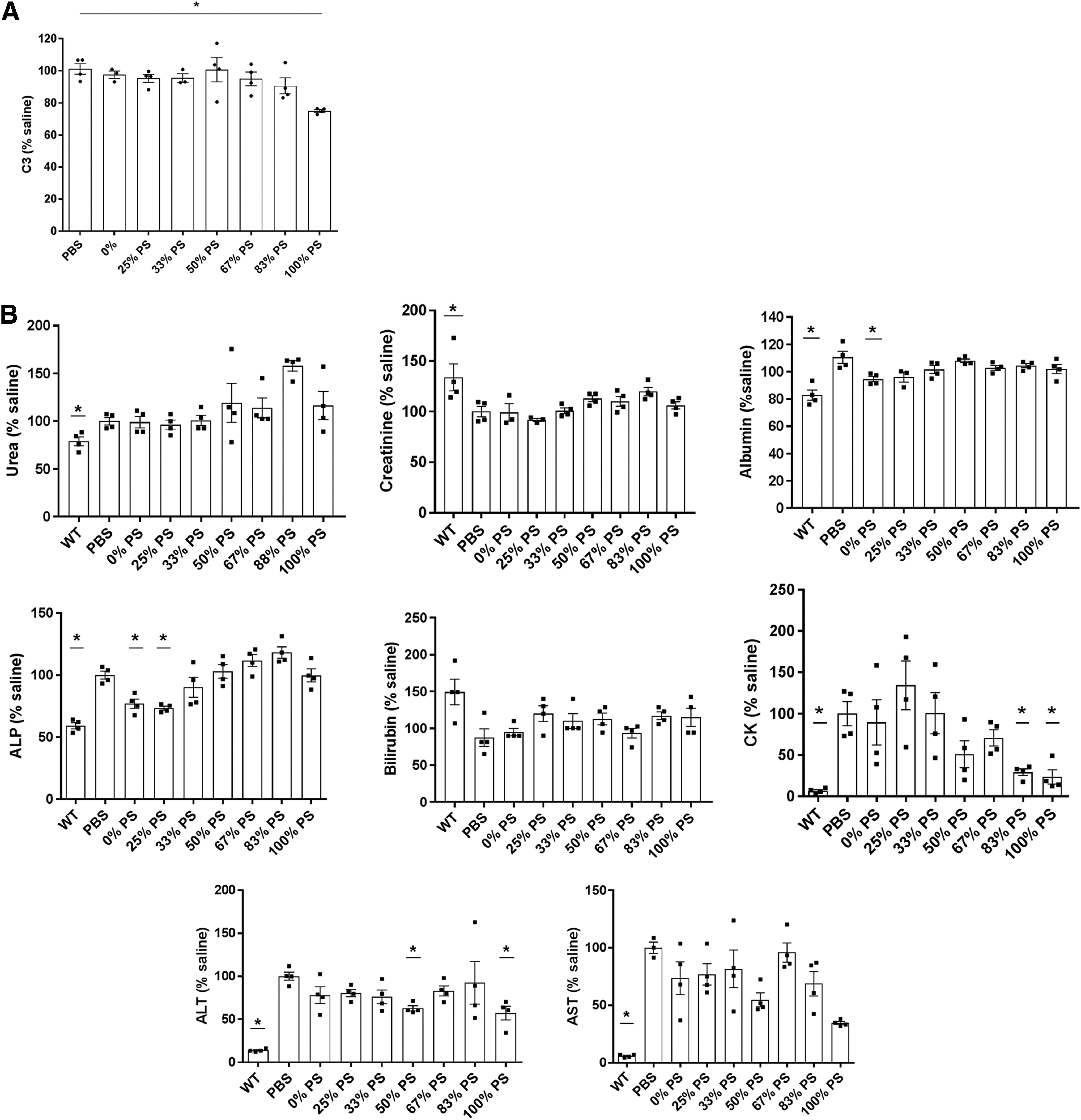
PS-modified tcDNAs show no associated toxicity after 4 weeks of treatment.
At the end of the 4-week treatment, sera were collected from treated mice to analyze various general biomarkers of toxicity (Fig. 5b). We quantified serum levels of urea, creatinine, albumin, ALP, bilirubin, and transaminases (ALT and AST) and observed no significant upregulation compared with mdx control mice (Fig. 5b). These results confirm previous work where hepatic toxicities have been primarily related to locked nucleic acid modifications rather than PS modifications [26]. We also quantified the serum CK level, a marker for muscle injury, which appears largely reduced in mice treated with tcDNA carrying 50% of PS bonds or more, confirming the efficacy of the treatment to restore dystrophin and improve the dystrophic pathology of the mdx mice (Fig. 5b). Similar decreases reflecting the treatment efficacy were also observed for AST and ALT levels.
Efficacy of the different PS compounds
To evaluate the contribution of PS content on tcDNA-ASO efficacy, we analyzed the levels of exon 23 skipping after 4 weeks of treatment with the 7 ASOs. Total RNA was extracted from various tissues, and levels of exon 23 skipping were analyzed by RT-PCR and quantified by RT-qPCR. The RT-PCR results presented in Fig. 6a reveal a skipped product in all tissues analyzed following treatment with all tcDNA-ASOs. Quantification clearly indicates that exon 23 skipping efficacy correlates with the number of PS linkages within the tcDNA-ASO (Fig. 6b). This is true for all analyzed tissues except the heart, where surprisingly the number of PS modifications does not seem to significantly impact exon skipping levels. For all the other muscle tissues analyzed (tibialis anterior, gastrocnemius, quadriceps, triceps, and diaphragm), there is a significant difference between the levels induced with the full PO compared with the full PS-tcDNA, correlating with the amount of compounds quantified in these tissues. Interestingly, while 25% and 33% PS compounds tend to induce similar exon skipping levels as the full PO, we found a substantial increase from 50% PS, suggesting that 50% of PS content may be sufficient to produce exon skipping levels similar to those of full PS-tcDNA (Fig. 6b).
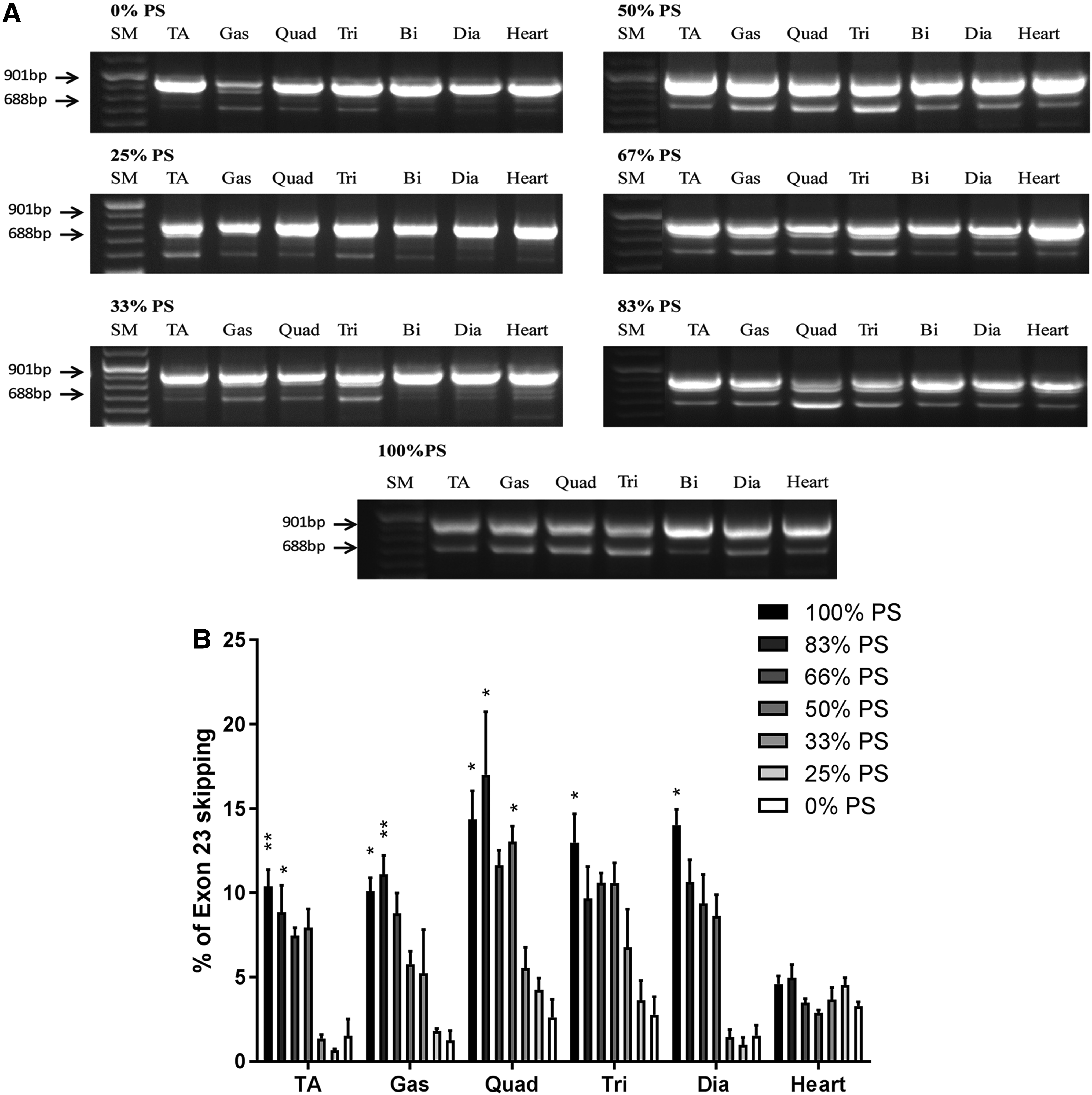
The number of PS modifications correlates with exon 23 skipping efficacy in all analyzed tissues of mdx mice, except in the heart.
We also analyzed the levels of dystrophin restoration in mice treated with ASOs carrying 0%, 50%, 67%, 83%, and 100% of PS modifications by WB (Fig. 7). Significant amounts of dystrophin protein (ranging from 1% to 15%) were detected in all analyzed tissues after only 4 weeks of treatment. Dystrophin levels follow the same trend as the one observed at the mRNA levels and appear to increase with the number of PS. We found particularly high levels of dystrophin restoration in the triceps (14.4% and 9.3%) and gastrocnemius muscles (8.2% and 6%) of animals treated with compounds carrying 83% and 100% of PS linkages compared with WT. We also confirmed low restoration levels of dystrophin protein in the heart of mice after 4 weeks of treatment, independently of the PS content.
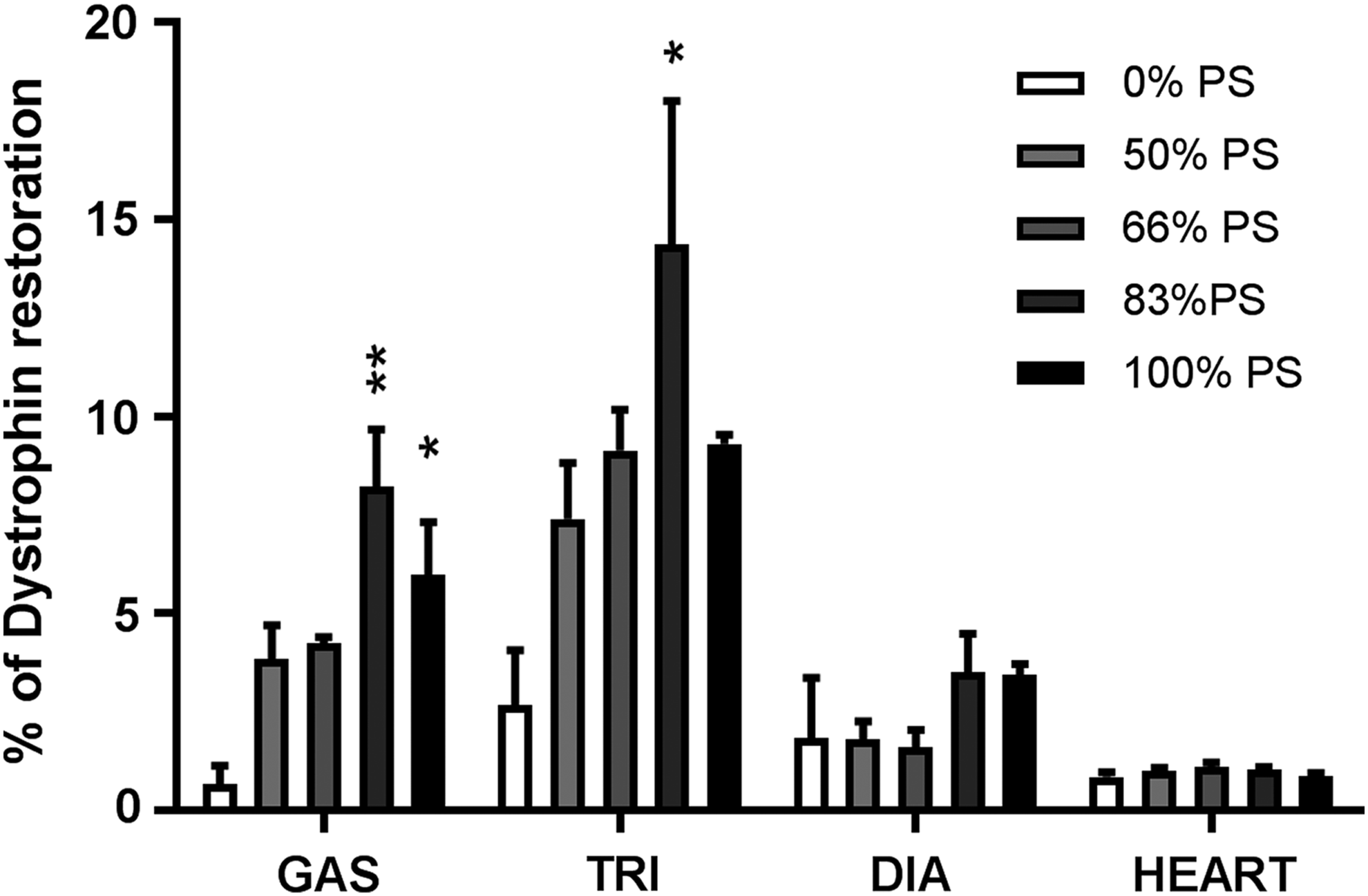
The amount of PS modifications correlates with dystrophin rescue in mdx mice muscle, except in the heart. Dystrophin restoration in mdx muscles was quantified by WB after systemic delivery (200 mg/kg/week) for 4 weeks of PS-modified tcDNA carrying 0% PS, 50% PS, 67% PS, 83% PS, and 100% PS. Results are expressed as mean ± SEM (n = 4 mice per group). *P < 0.05, **P < 0.01, compared with tcDNA 0% PS.
Based on biodistribution data and efficacy levels, we calculated the ratio of the exon skipping levels/tcDNA quantity per tissue to evaluate the therapeutic index of each compound, that is, to determine which compound produces the highest levels of exon skipping with minimal tcDNA accumulation. These ratios were calculated for the quadriceps, triceps, diaphragm, and heart and are reported in Table 2. The highest ratios were obtained for compounds containing 50% and 67% of PS. We also averaged the ratios obtained for all four muscles and plotted them in Fig. 8. These ratios averaged on the various muscle tissues indicate that tcDNAs carrying 50% or 67% of PS linkages present the highest efficacy/tcDNA quantity ratios (Fig. 8, Table 2).

Ratio of efficacy over tcDNA quantity in tissues. Efficacy over tcDNA quantity ratios was calculated in skeletal muscles (quadriceps, triceps, diaphragm, and heart) of mice injected with PS-modified tcDNA carrying 0% PS, 50% PS, 67% PS, 83% PS, and 100% PS.
Ratio Efficacy/tcDNA Quantity
Discussion
ASO-based therapy has demonstrated encouraging results for a variety of genetic disorders, which have led to several clinical programs currently ongoing. Most of these trials involve PS-modified ASOs, the most widely used modification for nucleic acid drug development, which helps the wide biodistribution of ASOs. Nevertheless, improving the delivery and safety is still the major challenge faced by the antisense platform. Among new promising chemistries, we have previously shown that tcDNA ASOs display interesting properties and clear superiority compared with clinically tested ASOs (2′OMe and PMO) for DMD [16]. It is important to note that one of the main characteristics that make tcDNAs particularly interesting is that they are stable in their full PO version as opposed to other chemistries, which require PS bonds for stability [18,19].
Beyond nuclease resistance, PS linkages have shown advantageous PK properties compared with unmodified phosphorodiester backbones, which have a very limited efficacy and are rapidly filtered and excreted into the urine due to poor plasma protein binding. On the contrary, fully modified PS oligonucleotides benefit from an extensive binding to plasma proteins, facilitating tissue uptake and preventing renal clearance.
While there are a vast number of studies on fully modified PS compounds [9,10,27], studies on how the number of PS modifications may contribute to ASO efficacy are rare [28–30]. To better characterize the contribution of PS linkages in tcDNA efficacy, this study compares the protein binding and exon skipping ability of seven tcDNAs targeting the exon 23 of mouse DMD gene and carrying different amounts of PS modifications (0%, 25%, 33%, 50%, 67%, 83%, and 100%). Our protein binding experiments demonstrate that the quantity of plasma proteins that binds to the tcDNA depends on its number of PS linkages and that it occurs independently of the ASO chemistry or its sequence.
PS ASOs have been associated with an increased risk of acute toxicity due to hybridization-independent mechanisms often restricted to high doses [9,31,32]. Among undesirable effects, inhibition of coagulation has been described as a class effect of PS-modified compounds associated with high drug plasma concentrations [33]. As expected, the results on the evaluation of coagulation parameters in mouse plasma in vitro showed increased PT and aPTT with increased number of PS linkages; aPTT being the most sensitive to the presence of PS. Interestingly, decreasing the number of PS had a positive impact by reducing the effect of ASOs on coagulation pathways. We also looked at the complement activation pathway, which has also been associated with PS-ASO toxicity. Most studies tend to investigate complement activation in monkey or human serum [32]. However, we previously found a correlation between some toxic tcDNA-ASOs and complement activation in mice (unpublished data and Supplementary Fig. S3). We therefore included complement activation screening in our mouse preclinical study. Here, none of the tcDNA compounds (being full PO or full PS) activated the complement in vitro in mouse or human serum. In addition, the evaluation of C3 levels in mouse serum 1 h after the first injection of the different PS-tcDNA compounds did not show any difference when compared with saline controls, except a slight decrease of C3 after the injection of 100% full PS-tcDNA, which was not associated with any clinical sign of toxicity.
In addition to acute complications, the increased protein binding properties of PS-modified oligonucleotides prevent their excretion in the urine as they form larger complexes that diminish their filtration by the glomerulus. Following 4 weeks of repeated systemic administration of tcDNA carrying different amounts of PS (from 0% to 100%), we observed a typical accumulation in untargeted organs such as the kidney and liver, in particular with compounds containing 50% of PS and above. The evaluation of hepatic (ALT, ALP, AST, bilirubin) or renal injury (urea, creatinine, albumin) biomarkers in mouse serum at the end of the treatment did not reveal significant upregulation in treated animals compared with their saline controls. However, longer treatments should be performed to evaluate more thoroughly repeated dose-associated toxicity.
In this study, we focused on the impact of the number of PS linkages on tcDNA biodistribution and exon skipping efficacy. Following systemic IV administration, the serum levels of full PO (0%) and full PS (100%) tcDNAs rapidly declined from a peak concentration followed by a long terminal elimination phase, as previously described for other ASOs [23]. This apparent long elimination rate has previously been explained by the slow removal of ASOs from tissues in the postdistribution phase, creating an equilibrium of ASO concentration between plasma and tissues [22]. As expected, full PS-tcDNA shows an advantageous PK profile compared with full PO-tcDNA, however, their PK profiles were not different enough to justify the study of plasma concentrations of intermediate PS-tcDNA compounds. Both compounds are rapidly distributed from blood circulation; however, their fate appears to be different. PO-tcDNA may indeed have a higher renal clearance due to the poor protein binding, while full PS-tcDNA may have more effective biodistribution and cellular uptake. This is in concordance with biodistribution results, which indicate that tcDNA amounts in muscle tissues as well as in the kidney and liver are extremely dependent on PS content and are particularly high in full PS-tcDNA-treated tissues, as shown for other PS-modified chemistries. Accordingly, exon skipping efficacy and dystrophin restoration levels were found to be higher with increased number of PS in all tested tissues except in the heart. The cardiac muscle has indeed been shown to be more challenging to target and may involve particular uptake mechanisms. It has been postulated that dystrophin-negative muscle fibers become “leaky,” facilitating ASO uptake, but cardiomyocytes do not share this “leaky” condition and are therefore more difficult to target [34,35].
Interestingly, we found that tcDNA with 50% and 67% of PS content induces similar levels of exon 23 skipping compared with the full-PS counterpart, although fuller PS-tcDNAs were quantified in these tissues. Indeed, while the accumulation of tcDNA in tissues appears dependent on PS content, it is not directly proportional and increases dramatically from a threshold of 83% PS and even more with 100% PS. Calculation of efficacy/tcDNA quantity per tissue therefore indicates that tcDNA compounds carrying 50%–67% of PS links present the highest therapeutic index (i.e., producing similar exon skipping levels with less tcDNA accumulation). These observations raise an interesting question as to why higher amounts of ASOs in tissues do not lead to higher efficacy. We hypothesize that this may be due to a difference in productive uptake. Similar exon skipping levels suggest similar levels of productive uptake, that is, delivery to the nuclei, whereas tcDNA quantification in tissues reflects general uptake (both productive and unproductive). These results therefore suggest that high PS content such as 83% or 100% leads to high tissue accumulation, without necessarily being productive uptake.
Altogether, our results indicate that from 50% to 67% of PS content, tcDNA-ASOs induce similar levels of exon 23 skipping compared with the full-PS counterpart with less unproductive accumulation. These findings support the possibility of reducing PS content, which may result in lower long-term toxicity, without affecting significantly the efficacy. These data contribute to further understand the contribution of PS linkages in antisense efficacy and will help the design of future safe and efficient tcDNA-ASOs for clinical studies.
Footnotes
Acknowledgments
We thank Branislav Dugovic, Reto Bertolini, Thomas Tensorer, and Patrick Lûthi from SYNTHENA, Berne, Switzerland, for providing the tcDNA-ASOs used in this study. We also thank John-Paul Carter for proofreading the article. This work was supported by the Agence nationale de la recherche (Chair of Excellence HandiMedEx), the Institut National de la santé et la recherche médical (INSERM), the Association Monegasque contre les myopathies (AMM), the Duchenne Parent Project France (DPPF), the Fondation UVSQ, and by the Fondation pour la Recherche Médicale, FRM grant no. PLP20161036676 to P.A.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.