
Abstract
Huntington's disease is a neurodegenerative disorder caused by a CAG repeat expansion in the first exon of huntingtin gene (HTT) encoding for a toxic polyglutamine protein. This disease is characterized by motor, psychiatric, and cognitive impairments. Currently, there is no disease modifying treatment. However, reducing the expression of the huntingtin protein (HTT) using antisense oligonucleotides (ASOs) has been shown as a promising therapeutic strategy. In this study, we explore the therapeutic potential of ASO made of tricyclo-DNA (tcDNA), a conformationally constrained DNA analog, to silence HTT. We used a gapmer ASO, containing central DNA nucleotides flanked by tcDNA modifications on 5′ and 3′ ends, allowing the recruitment of RNAse H and subsequent degradation of the messenger RNA. After transfection of tcDNA-ASO in patient-derived fibroblast cell lines, we show a strong decrease of HTT mRNA and protein levels. As a control, 2′O-methyl-RNA targeting the same region of HTT was also tested and did not induce a significant effect. tcDNA-ASO were also evaluated in vivo in the YAC128 mice, containing the full-length human HTT gene with 128 CAG repeat expansion. Single intracerebroventricular (ICV) injections of tcDNA induce a significant decrease of HTT messenger and protein levels in the cortex, hippocampus, striatum, and cerebellum of treated mice. tcDNA-ASO were found well distributed in the central nervous system (CNS) and show long lasting effect with protein levels still low, 12 weeks after a single ICV injection. This proof of concept study suggests the therapeutic potential of gapmer tcDNA ASO to downregulate huntingtin in vitro and in vivo.
Introduction
Huntington's disease (HD) is an autosomal dominant disease characterized by neurodegeneration of striatum, which leads to motor, cognitive, and behavioral problems. It is caused by CAG expansions in the exon 1 of HTT gene [1], encoding an elongated polyglutamine tract in the huntingtin protein (HTT). HTT is a 348 kDa protein ubiquitously expressed [2] even if the striatum is primarily affected in HD. Patients also present peripheral manifestations of the disease such as weight loss and an increase of peripheral inflammation [3]. Currently, there is no disease modifying treatment for HD and most therapies aim at ameliorating symptoms.
While the pathogenesis of HD is still unclear, ongoing research focuses on the primary cause of the disease, the mutant HTT protein. Gene silencing approaches have been tested using small interfering RNAs, microRNAs, or short-hairpin RNAs to achieve downregulation of HTT by RNA interference (RNAi) mechanism [4–7]. The use of antisense oligonucleotides (ASOs) has also been investigated to lower the levels of HTT. Gene silencing approaches for HD offer two main options: allele specific, which aims at knocking down specifically the mutant allele, or nonspecific silencing, which will lower both alleles and therefore all HTT protein. One way to induce a preferential silencing of the mutant allele is to directly target the CAG repeats, which are more represented on the mutant allele. Several types of ASOs have been tested, such as peptide nucleic acid, locked nucleic acid, 2′O-methyl (2′OMe)-RNA, or phosphoroamidate morpholino oligomer to target HTT mRNA [8–10], or more recently DNA [10 and 11], and have induced significant decrease of mutant HTT. However, the specificity for mutant HTT is still debated since CAG repeats are also present on wild-type allele, and most patients have a number of CAG repeats relatively close between both alleles.
More specific approaches using ASOs targeting polymorphisms have been used. In that case, gapmer ASOs enabling the recruitment and activation of RNase H are used to knock down the expression of the targeted mRNA. Carroll et al. have used a 2′O-methoxyethyl-RNA (2′MOE) gapmer ASO targeting a single-nucleotide polymorphism (SNP) present in 49% of HD patients and have shown strong and specific silencing of mutant HTT in an HD mouse model [12]. Other modifications such as S-constrained ethyl [13–15] have also been tested, and Southwell et al. have recently demonstrated that both early and late treatment reduced cognitive and behavioral impairments in mice [16], confirming the therapeutic potential of this approach. Nevertheless, allele-specific strategies face the challenges of personalized medicine, since no SNP was found common to the entire HD population [17].
An alternative strategy is the nonspecific silencing approach aiming at lowering the global level of huntingtin. This can be achieved with gapmer ASO targeting a sequence common to both alleles. Intracerebroventricular (ICV) infusions of 2′MOE gapmers during 2 weeks have shown an efficient and sustainable reduction of HTT levels and a phenotypic reversal in three different HD mouse models [18]. These encouraging preclinical results have led to the evaluation of this 2′MOE gapmer (IONIS HTTRx) in clinical trials (ClinicalTrials.gov Identifier: NCT02519036), in which the drug is directly injected in the spinal fluid (intrathecal administration) of HD patients. Recent press releases revealed that “dose-dependent reductions of mHTT were observed in participants treated with IONIS-HTTRx and that phase 1/2 study demonstrates correlation between reduction of disease-causing protein and improvement in clinical measures of Huntington's Disease.” As promising as these results are, it should be noted that HTT lowering was detected in the cerebrospinal fluid (CSF) of treated patients and it is not clear which part of the brain caused the reduced concentration and whether lowering huntingtin levels can halt or slow the progression of HD.
Because of the inability of most ASOs to cross the blood–brain barrier (BBB), current antisense treatment for HD must be administered via ICV or intrathecal route (whether being in preclinical studies or in trials). These ways of administration not only present obvious clinical challenges, especially when repeated injections are required, but also neglect the issue of delivering ASOs to the periphery, which could be crucial for HD [3]. In this context, developing ASO, which could deal with the issue of both peripheral and central tissue penetration, represents an interesting option.
Tricyclo-DNA (tcDNA) ASOs have demonstrated unique uptake properties and the ability to cross the BBB at low levels after systemic administration such as intravenous or subcutaneous delivery in various mouse models [19–22]. tcDNA are constrained DNA analogs displaying increased RNA affinity and stability toward nuclease degradation. Fully modified tcDNA containing all four natural nucleobases reveal increases in thermal stability of duplexes with complementary RNA of ca. 2.4°C/mod [23,24]. tcDNA ASOs have been used to modulate splicing in their fully modified form as a therapeutic strategy for Duchenne muscular dystrophy and spinal muscular atrophy (SMA) [19–22]. In this study, we wanted to investigate the therapeutic potential of tcDNA ASO under gapmer design to downregulate HTT mRNA via RNAse H activation. We first demonstrate the lowering of HTT levels in vitro in various fibroblasts from HD patients and then in vivo after ICV injections in YAC128 mice. This proof of concept study confirms the potency of gapmer tcDNA ASOs to silence HTT and opens future research perspectives investigating possibilities of a simpler mode of administration targeting both peripheral and central tissues.
Materials and Methods
Oligonucleotides
Two different gapmer-ASOs were used. tcDNA-ASO was 15 nucleotides long with four tcDNA modifications on the 5′ and 3′ ends (upper case letters), seven unmodified nucleotides in the central part (lower case letters) with phosphorothioate linkages (marked with an asterisk), and was synthesized by Synthena (Bern, Switzerland): 5′-C*A*G*T*a*a*c*a*t*t*g*A*C*A*C-3′. 2′OMe-ASO was 20 nucleotides long with five 2′OMe modifications on the 5′ and 3′ ends, 10 unmodified nucleotides in the central part with phosphorothioate backbone, and was synthesized by Eurogentec: 5′-C*U*C*A*G*t*a*a*c*a*t*t*g*a*c*A*C*C*A*C-3′. ASOs were resuspended in sterile phosphate-buffered saline (PBS). This sequence was previously described by Kordasiewicz et al. [18].
Cell culture and transfection
Patient-derived fibroblasts (GM02171-17/17 CAG; GM02147-15/43 CAG; GM03621-18/59 CAG and GM09197-18/180 CAG) were purchased from Coriell Institute (Camden, NJ). Cells were cultured in F10 medium with 20% fetal bovine serum and 1% penicillin/streptomycin at 37°C with 5% CO2. ASO transfection was performed with 12 μL of Lipofectamine 2000 (Life Technologies) and 3 μg of antisense in 2 mL in a six-well plate, according to the manufacturer's protocol. Cells were harvested 48 h after transfection for RNA analysis and 72 h for protein analysis.
Animals
Animal procedures were performed in accordance with National and European legislation, approved by the French government (Ministère de l'Enseignement Supérieur et de la Recherche, Autorisation APAFIS#6519-20J 6082316012087). YAC128 mice were obtained from Jackson Laboratories and were bred in our animal facility at the Plateforme 2Care, UFR des Sciences de la santé, Université de Versailles-Saint Quentin. Mice were maintained in a standard 12 h light/dark cycle with free access to food and water. Transgene positive males were crossed with wild-type (wt) females, producing offspring of 50% each phenotype. Tail tissue for genotyping was collected and DNA was extracted after 35 min at 98°C with NaOH 50 mM and neutralization with Tris-HCl 1 M pH 8,2. Specific primers for human HTT gene were used (forward primer: 5′-TGCCAGCACTCAAGAAGGACAC-3′ and reverse primer: 5′-CACGCCAAGAATCAGCAGAGTGG-3′), leading to a product of 749 bp if the mouse is transgene positive. Murine GAPDH was used as a positive control (forward primer: 5′-TGACGTGCCGCCTGGAGAAA-3′ and reverse primer: 5′-AGTGTAGCCCAAGATGCCCTTCAG-3′). Animals were housed in a clean animal facility under a 12 h day/night cycle, with free access to food and water. Mice were anesthetized using ketamine (90 mg/kg) and xylazine (10 mg/kg). ASOs were stereotaxically injected into the right lateral ventricle through a needle connected to a Hamilton syringe at a rate of 0.5 μL/min. The bregma coordinates were 0.2 mm anterior; 1 mm lateral, and 2.5 mm depth. The needle was left in place for 5 min then slowly withdrawn. Animals were euthanized 2, 6, or 12 weeks after injection. Brains were removed, divided into right and left hemisphere, and cortex, striatum, hippocampus, and cerebellum were isolated and snap frozen in liquid nitrogen.
RNA isolation and reverse transcription-quantitative polymerase chain reaction
Cells were harvested 48 h after transfection, and Total RNA was extracted with TRIzol reagent and transferred into columns (Kit NucleoSpin RNA; Macherey Nagel) with a DNAse treatment. cDNAs were obtained from 0.1 to 1 μg of RNA with SuperScript III First-strand synthesis system (ThermoFisher) using random hexamer primers. Quantitative PCR (qPCR) was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) and 300 nM of each primer and analyzed in CFX96 Real-Time PCR (Bio-Rad). HTT mRNA levels were assessed with two pairs of primers (Forward_exon7: 5′-TGCCAGCACTCAAGAAGGACAC-3′; Reverse_exon8: 5′-CACGCCAAGAATCAGCAGAGTGG-3′; Forward_exon64: 5′-CGACAGCGAGTCAGTGATTG-3′; Reverse_exon65: 5′-ACCACTCTGGCTTCACAAGG-3′) and normalized with human GAPDH for in vitro analysis (Forward_GAPDHhum: 5′-GAGTCAACGGATTTGGTCGT-3′; Reverse_GAPDHhum: 5′-TTGATTTTGGAGGGATCTCG-3′) or with murine GAPDH for in vivo analysis (Forward_GAPDHmur: 5′-TGACGTGCCGCCTGGAGAAA-3′; Reverse_GAPDHmur: 5′-AGTGTAGCCCAAGATGCCCTTCAG-3′).
Protein isolation and immunoblotting
Proteins were extracted with Pierce radioimmunoprecipitation assay (RIPA) buffer (ThermoScientific) and 1 × complete protease inhibitor (Roche). Protein concentrations were determined with Pierce BCA protein assay kit (ThermoScientific). Samples were prepared with 4 × NuPAGE lithium dodecyl sulfate Sample buffer and 10 × Sample reducing agent (Life Technologies) and heated at 70°C for 10 min. Ten to thirty micrograms of total proteins were loaded in a 3%–8% NuPAGE Tris Acetate precast gels (Invitrogen) with Tris Acetate Running Buffer at 80 V for 2 h and 110 V for 3 h. Proteins were transferred to nitrocellulose overnight at 100 mA in a cold room in Transfer Buffer with 10% ethanol. Membranes were then blotted with anti-HTT [MAB 2166 (total HTT) or MAB1574 (anti-polyglutamine expansion) 1:1,000; Millipore] and normalized with antivinculin (Sigma). Proteins were detected with IR Dye 800 goat anti-mouse (Licor; 1:2,000) using the Licor Odyssey system for scanning the membrane.
tcDNA quantification by fluorescent hybridization assay
Tissues were homogenized in proteinase K buffer (100 mmol/L Tris–HCl, pH 8.5, 200 mmol/L NaCl, 5 mmol/L ethylenediaminetetraaceticacid, 0.2% sodium dodecyl sulfate) containing 2 mg/mL of proteinase K (Invitrogen) (50 mg tissue/mL of buffer), followed by incubation overnight at 55°C in a hybridization oven. After centrifugation at 14,000 rpm (Sorvall ST 8R centrifuge, 75005719 rotor) for 15 min, the supernatant was used in the assay. Quantification of tcDNA was performed using a hybridization assay with a fluorescently labeled DNA probe (Alexa680) after electrophoresis on acrylamide gel. In brief, tissue lysates were incubated 5 min at 50°C with a 5′ labeled (Alexa680) DNA complementary probe (Eurogentec) and run on a 15% acrylamide gel for 2 h at 110 V. The gel was imaged using the Licor Odyssey system and the amount of tcDNA in tissues was determined using a standard curve built on the measurement of known tcDNA quantities dissolved in the respective tissue lysates of mock-injected animals.
Statistical analysis
Data were analyzed by GraphPad Prism5 software (San Diego, CA) and shown as means ± standard errors of the mean. “n” refers to the number of mice per group. Comparisons of statistical significance were assessed by two-way analysis of variances followed by Bonferroni's post hoc tests for more than two group comparisons. Comparisons of statistical significance were assessed by nonparametric Mann–Whitney U tests. Significant levels were set at *P < 0.05, **P < 0.01, ***P < 0.001.
Results
tcDNA-ASOs silence HTT in HD fibroblasts
To assess the potency of tcDNA-ASO to downregulate the human huntingtin mRNA and protein levels, we designed a phosphorothioated tcDNA gapmer of 15-nt with four tcDNA modifications on 5′ and 3′ ends flanking a 7-nt DNA core, targeting a sequence in exon 36 of the human HTT gene. We compared the efficiency of this tcDNA gapmer with a 2′OMe-RNA gapmer of 20-nt made of a central 10-nt DNA core flanked with two wings of 5-nt 2′OMe. The efficacy of these ASOs was first assessed in vitro by transfection with Lipofectamine in HD fibroblasts carrying various CAG repeats. Forty-eight hours after transfection, cells were harvested and the effect of the ASO on RNA level was evaluated by reverse transcription polymerase chain reaction (RT-PCR) around the CAG repeats to distinguish between both alleles. As represented in Fig. 1A, showing a representative gel from transfection in HD fibroblasts carrying 59/18 CAG repeats, two bands are detected; the upper band corresponds to the mutant allele, and the lower band to the wt allele. Both alleles appear reduced after tcDNA treatment confirming the nonspecific downregulation induced by this gapmer ASO, whereas the 2′OMe did not induce any silencing. To quantify more accurately the levels of downregulation, total HTT mRNA was assessed by qPCR around exons 64–65 (Fig. 1B). Relative quantification was performed using GAPDH as a reference and compared to control conditions (set at 100%). In four different cell lines (one wt and three HD patient lines), tcDNA-ASO significantly decreased the levels of human huntingtin mRNA to 40%–30% of the control levels, independently of CAG repeat numbers. We then assessed the huntingtin protein levels by western blot in the GM09197 HD fibroblasts carrying 180/18 CAG, enabling a proper separation of mutant and wild-type proteins (Fig. 1C). MAB2166 antibody, which is specific of HTT protein, recognizes both forms, whereas MAB1574 is specific of the polyglutamine expansion, identifying only mutant HTT protein. Vinculin was used as a loading control. The western blot results show a decrease of both mutant (Fig. 1D) and wt (Fig. 1E) HTT protein level to 80% and 30% of control levels after 2′OMe-ASO and tcDNA-ASO, respectively, confirming a strong silencing with tcDNA in GM09197 (180/18CAG) cell line. In other cell lines carrying less CAG repeats, total HTT protein levels were also reduced to 40% to 20% of control levels after tcDNA treatment, while 2′OMe-ASO only induced a marginal decrease (Supplementary Fig. S1).
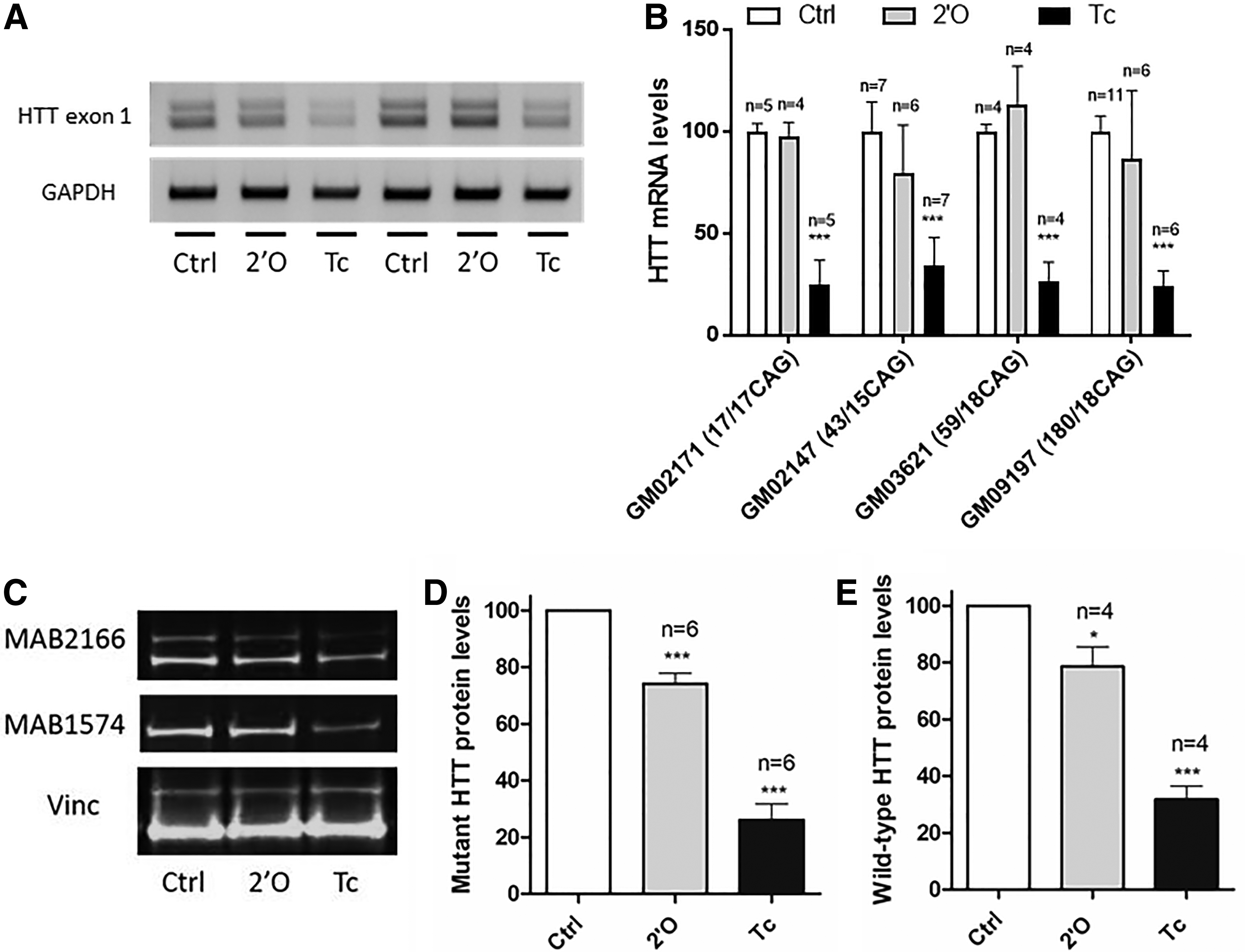
Evaluation of tcDNA-ASO efficiency after transfection in HD patient-derived fibroblasts.
tcDNA-ASOs downregulate HTT in YAC128 mice
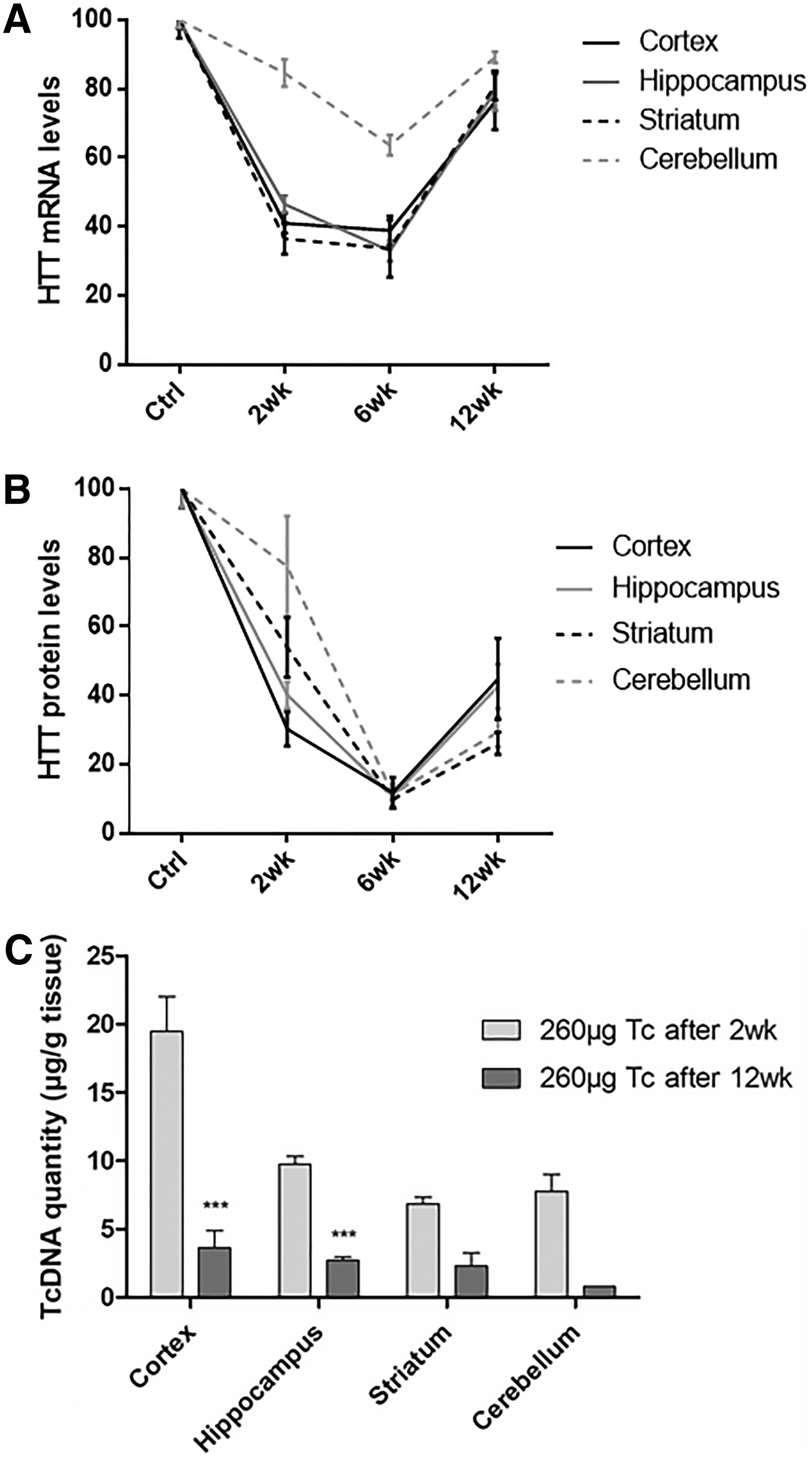
To evaluate the effect of tcDNA-ASO on HD in vivo, we used the transgenic YAC128 mouse model, carrying the entire human HTT gene with 128 CAG repeats in addition to its endogenous Hdh gene. The mice received a single ICV injection of 130 or 260 μg of tcDNA and were compared to PBS-injected mice. Interestingly, these doses of tcDNA were well tolerated in YAC128 mice, whereas equimolar doses of 2′OMe were lethal for the mice. Brains from injected mice were isolated 2 weeks after treatment and cortex, hippocampus, striatum, and cerebellum were analyzed. qPCR around exons 64–65 shows a significant decrease of HTT mRNA to 40% of the level of PBS controls in cortex, hippocampus, and striatum of tcDNA injected mice with the two different doses (Fig. 2A). Similar results were obtained with a qPCR around exons 7–8 (Supplementary Fig. S2). The levels of mutant HTT protein were assessed by western blot and were found significantly decreased in all four central nervous system (CNS) regions analyzed. The silencing of the human mutant huntingtin was particularly strong in the cortex with levels down to 30% of PBS controls (Fig. 2B), which correlates with qPCR results. The endogenous wt murine HTT was also quantified and was not affected as expected from the specificity of the tcDNA-ASO for human HTT and not murine Hdh (Supplementary Fig. S3).

Evaluation of tcDNA efficacy after ICV injection of 130 or 260 μg of tcDNA in YAC128 mice.
Interestingly, the 260 μg dose induced only a slightly stronger silencing compared with the 130 μg. To correlate the silencing levels with ASO amount and evaluate the biodistribution of the tcDNA-ASO following direct ICV injection, we measured the amounts of tcDNA-ASO in the various brain regions isolated (Fig. 2C). tcDNA-ASO were effectively detected in all four CNS regions with no significant difference between the 260 μg injected group and the 130 μg injected mice. tcDNA-ASO distribution was not uniform in all four brain regions analyzed with significantly higher amounts in the cortex (17–20 μg), correlating with the levels of silencing observed.
Long-lasting effect of tcDNA-ASO
We then investigated the extent and duration of the mutant huntingtin silencing over time following a single ICV injection of tcDNA-ASO. Six weeks after the injection of 260 μg of tcDNA-ASO, the level of human huntingtin mRNA appears lower than levels detected 2 weeks after the injection. qPCR reveals a decrease of HTT mRNA down to 38%, 32%, and 33% of the level of PBS controls in cortex, hippocampus, and striatum, respectively (Fig. 3A). Similarly, the silencing of the human muHTT is also stronger with levels down to ∼10% of PBS controls in all four CNS regions (Fig. 3B, C).

Sustained effect of tcDNA 6 weeks after 260 μg of tcDNA.
We also assessed the silencing levels 12 weeks after a single ICV injection and found HTT mRNA levels coming back up, with a decrease to only 75%–80% of PBS controls (Fig. 4A). Interestingly, HTT protein levels were still low (down to 26%–44% of PBS controls) (Fig. 4B, C), suggesting the long-lasting effect of tcDNA to silence HTT (Fig. 5A, B). The kinetic of the mRNA and protein silencing summarized in Fig. 5A and B clearly shows a stronger and longer silencing at the protein level.
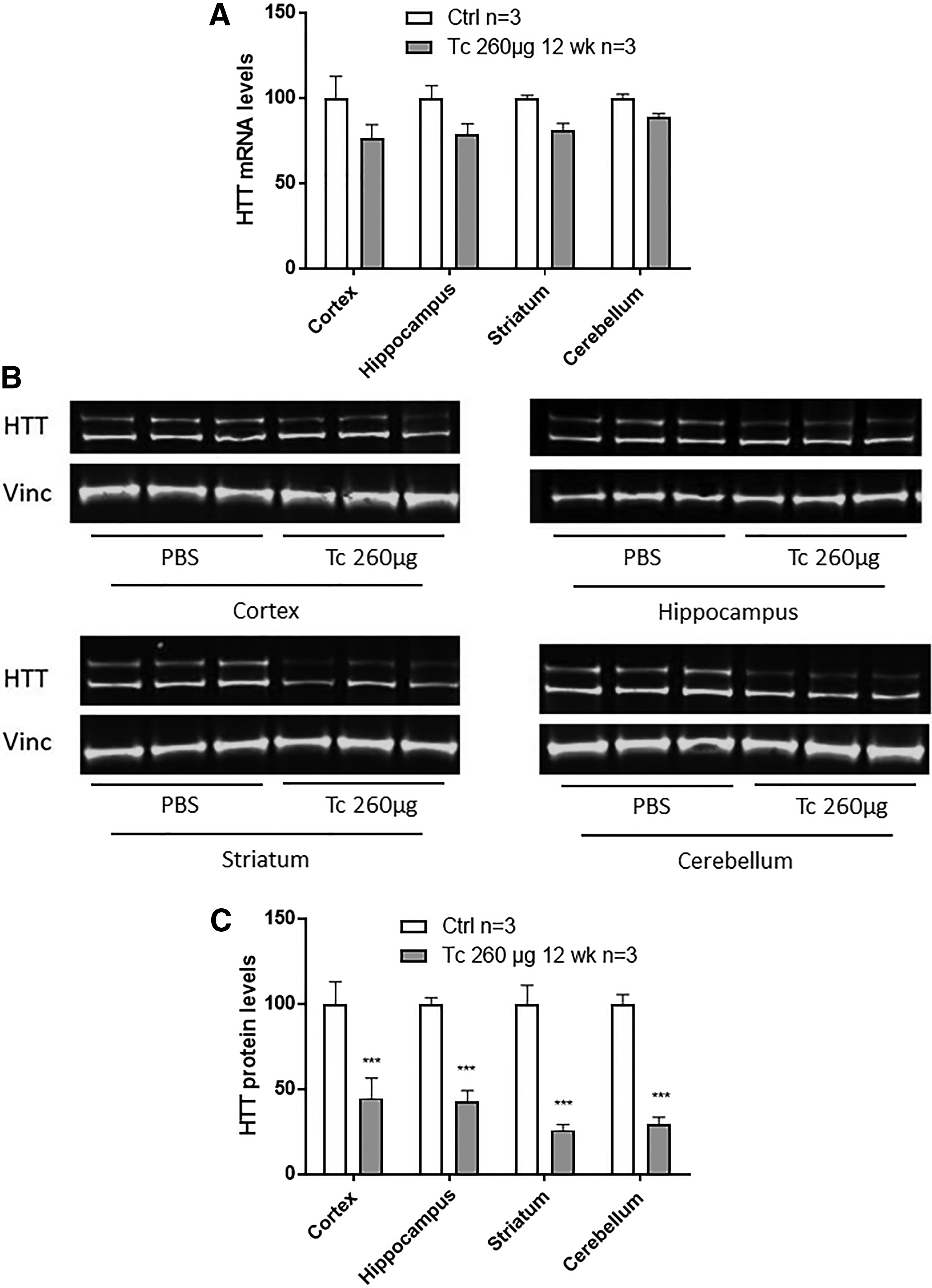
Sustained effect of tcDNA 12 weeks after 260 μg of tcDNA.
Long-lasting effect of tcDNA in brain after ICV injection.
To correlate these silencing levels to the amounts of ASOs, we measured the amount of tcDNA in the brain 12 weeks after the injection (Fig. 5C). tcDNA-ASOs were still detected in all four CNS regions analyzed 12 weeks after a single ICV injection but at much lower levels than 2 weeks after (<5 μg of tcDNA/μg tissue). Across the four brain regions, amounts of tcDNA-ASO detected 12 weeks postinjection were found ∼5 times lower than the amounts detected 2 weeks after.
Discussion
Our results demonstrate that tcDNA-ASO can successfully downregulate HTT in HD fibroblasts carrying various number of CAG repeats as well as in vivo in YAC128 mice after a single ICV injection. Following direct injection in the cerebral ventricle, tcDNAs were found in various regions of the CNS, including cortex, striatum, hippocampus, and cerebellum. tcDNA-ASO distribution appeared nonuniform with higher amounts of ASOs detected in the cortex, which might be a result of the slow injection protocol used. Previous studies have compared slow and fast ICV delivery and shown more efficient and uniform distribution after fast delivery [16,25]. The broad distribution of the ASO is particularly important for the treatment of HD, which is not a pathology restricted to the striatum. Huntingtin is ubiquitously expressed and while the striatum is particularly vulnerable to mutant huntingtin toxicity, collective evidence suggests that effective treatment for HD will require targeting multiple brain regions [26]. This widespread distribution represents an advantage of single-stranded ASOs compared with double-stranded RNA displaying rather low diffusion and cellular uptake in the CNS, therefore limiting the therapeutic potential of RNAi approaches. However, recent years have seen the development of enhanced delivery system and many serotypes of viral vectors such as adeno associated virus, which can induce widespread diffusion and long-term treatment [26].
In this study, we evaluated two doses of tcDNA-ASO, 130 and 260 μg, which were well tolerated in YAC128 mice. In contrast, similar doses of 2′OMe ASO were toxic and treated mice did not survive the injection, which disallowed the comparison between the two ASOs. Surprisingly, we did not find a much stronger silencing in mice treated with 260 μg of tcDNA than in the 130 μg group. These results correlate with the amount of ASO quantified in tissues 2 weeks after the injection, which were not significantly different between the two groups. As opposed to previous studies using an infusion of ASO over 2 weeks leading to a total amount of up to 700 μg of 2′MOE-ASO injected [18], we performed a single ICV injection, therefore corresponding to lower amount of ASO. Nevertheless, the levels of HTT lowering obtained with tcDNA-ASO were similar to those obtained with higher dose of 2′MOE. The maximum silencing effect was detected 6 weeks after administration with HTT levels measured as low as 10% of control levels. While the downregulation of HTT mRNA appears to diminish 12 weeks after injection, protein levels were still low (down to 28% of control levels in the striatum). tcDNA-ASOs were still detected in the various brain regions 12 weeks after a single injection, suggesting a long-lasting effect of tcDNA. Amounts measured 12 weeks after a single ICV were ∼5 times lower than levels detected 2 weeks after the injection, which indicates a slightly lower half life than fully modified tcDNA-ASO previously described [19]. This can be expected considering the core of unmodified DNA nucleotides within the gapmer ASO, which likely facilitates its degradation. However, these results suggest that low amounts of oligonucleotides (<5 μg/g of tissue) can still induce some level of downregulation. More importantly, it was previously shown that the phenotype rescue is maintained even when the mutant protein level is going back to pathological level [18]. This important observation, also named “huntingtin holiday” indicates that transient suppression of mutant huntingtin can have a prolonged effect on the disease. While the objective of the current study was to establish the proof of concept of tcDNA-mediated downregulation of HTT, it would be interesting to assess the impact of tcDNA gapmers on the phenotype of various mouse models and determine how long the advantageous effect can last.
Moreover, since tcDNA-ASOs have been shown to cross the BBB at low levels following systemic injections, they could represent an attractive tool to tackle both central and peripheral HTT-mediated toxicity. While HD presents a predominant neuropathological signature, phenotypes have been observed in a wide range of organ systems [3].
Current ways of treatment administration (ie, intrathecally) may not only raise concerns about giving lumbar punctures to patients on a regular basis, potentially for decades, but also neglect the peripheral contribution of the disease. Understanding the link between CNS manifestations of the disease and the peripheral causes of death remains extremely important. For example, questions about the cardiac dysfunction being secondary to neurodegeneration in the CNS are still unanswered and could lead to cruel outcomes if CNS-targeted therapies turned out unable to rescue potential detrimental consequences of having mutant HTT in peripheral organ systems [3]. However, this might never be an issue as a recent study investigating the peripheral silencing of huntingtin showed no amelioration of central signs of the disease in a mouse model of HD [27], highlighting once again the current debate about peripheral contribution in HD pathogenesis.
tcDNA could be particularly interesting to investigate such an influence and could become an attractive alternative antisense therapy for HD treatment. Nonetheless, systemic delivery of tcDNA such as other ASOs leads to accumulation in liver and kidneys. Considering the current low BBB penetrance of tcDNA, high doses of ASOs would be required to achieve substantial HTT lowering in the brain and this could raise unnecessary safety concerns. Beyond the typical proinflammatory/immunostimulatory effects associated with systemic administration of ASO (particularly those of the phosphorothioate [PS] class), detrimental effects could also emerge from high levels of peripheral HTT lowering, in particular, with a nonselective HTT-lowering compound.
In this study, we focused on a nonallele-specific silencing of HTT where the gapmer ASO is designed to downregulate both alleles and therefore the global levels of HTT protein. However, the safety of this strategy is still under debate since the wt huntingtin is also downregulated. The function of huntingtin is essential in early development, but is unclear in adulthood. Different studies have demonstrated that lowering both normal and mutant huntingtin was well tolerated in rodents and nonhuman primates [4,18, and 28]. Nevertheless, the effects of lowering huntingtin over a prolonged period of time are still unknown. The results of on-going and future trials will be crucial to determine whether the beneficial effects of lowering the toxic mutant huntingtin protein can outweigh any potential detrimental effects of lowering the wt huntingtin.
An intrathecally delivered ASO (HTTRx developed by Ionis Pharmaceuticals) that aims to lower global levels of huntingtin is now well into its first human clinical trial (ClinicalTrials.gov Identifier: NCT02519036), and two other ASOs (developed by Wave Life Science) aiming at lowering exclusively the mutant huntingtin have also entered phase I trials (ClinicalTrials.gov Identifiers: NCT03225833 and NCT03225846). These ASOs each target one of the two most common SNPs in mutated HTT and therefore represent a personalized route to treating HD since only patients with the targeted SNPs could benefit from the treatment. Nonetheless, the long-term results of these trials will definitely shed some light on the debated role of huntingtin in adults and the safety of a nonallele-specific approach.
It is now a pivotal time for antisense therapies with the first clinical success reported in 2016, when the United States Food and Drug Administration approved nusinersen for the treatment of SMA. Nusinersen is an intrathecally delivered ASO aiming at modulating the splicing of the survival motor neuron 2 (SMN2) gene to restore the expression of SMN protein. One could argue that the therapeutic benefit may be more easily demonstrated when rescuing a missing protein in the context of a particularly severe condition such as type I SMA compared to lowering the expression of a toxic protein such as HTT. However, these promising clinical results demonstrate the ability of an antisense oligonucleotide to slow down neurodegeneration in patients and therefore raise high expectations for similar strategies in HD.
Despite the great progress in the antisense field, one of the main challenges remains is the delivery of the ASO. Neurodegenerative diseases such as HD are of particular interest since ASO can be directly injected into the brain but improved delivery and ASOs should be considered and investigated. This proof of concept study demonstrates that tcDNA-gapmers could represent an alternative tool to the existing ASO to lower HTT levels.
Footnotes
Acknowledgments
We thank SYNTHENA, Berne, Switzerland, for providing the tcDNA-ASOs used in this study. This work was supported by the Agence nationale de la recherche (Chair of Excellence HandiMedEx), the Institut National de la santé et la recherche médicale (INSERM) and the Fondation UVSQ.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.