
Abstract
Dysferlinopathies encompass a spectrum of progressive muscular dystrophies caused by the lack of dysferlin due to missense mutations in the dysferlin gene or mutations causing premature truncation of protein translation. Dysferlin is a modular protein, and dysferlins lacking one or more repetitive domains have been shown to retain functionality. As such, antisense-mediated exon skipping has been proposed as a therapy for dysferlinopathy. By skipping the mutated exon, the reading frame would be maintained, while the mutation would be bypassed, thus allowing production of an internally deleted, but partially functional, dysferlin. We previously showed that dysferlin exon skipping is feasible in control cell lines. We here evaluated exon skipping and dysferlin protein restoration in patient-derived cells requiring the skipping of exon 9, 29, 30, or 34. Exon 30 skipping was possible at high efficiency, but did not result in increased dysferlin. We discovered that the alleged exon 30 mutation was in fact a polymorphism and identified a splicing mutation in intron 28 as the disease-causing mutation. While exon skipping was feasible for each of the other cell lines, no increases in dysferlin protein could be detected by western blotting.
Introduction
Dysferlinopathies consist of a spectrum of autosomal recessively inherited, progressive muscular dystrophies caused by the lack of the protein dysferlin. The most common dysferlinopathies are limb-girdle muscular dystrophy type 2B and Miyoshi myopathy [1,2]. Dysferlinopathies are very rare diseases with an estimated prevalence of 5–10 per million based on systemic reviews [3,4]. A recent effort from the Jain Foundation suggests an incidence of at least 8 per million and probably higher (Laura Rufibach, Jain Foundation, pers. comm.). Onset is usually in the second decade, with weakness in proximal extremities and, compared with other limb-girdle muscular dystrophy types, progression is relatively rapid. The disease is caused by mutations in the DYSF gene, mostly small deletions, insertions, and substitutions within exons, without an apparent mutation hotspot [1,2,5]. About half of all dysferlinopathy patients have a homozygous mutation, while the other half carry compound heterozygote mutations [6]. Mutations are distributed over the transcript without a hotspot.
Dysferlin is a modular protein that contains 6–7 Ca2+-dependent phospholipid-binding C2 domains and is involved in membrane repair [7,8]. It has been shown that dysferlins lacking one or more internal C2 domains maintain their membrane repair capacity [9]. In fact, a 70-year-old individual with very mild muscle weakness and elevated plasma creatine kinase has been reported to produce low levels of dysferlin lacking the fourth C2 domain [10]. In this individual, a DYSF null mutation abolished dysferlin production on one allele. On the other allele, a branch point mutation in intron 31 resulted in the skipping of in-frame exon 32 and production of 10% of wild-type levels of dysferlin lacking the amino acids encoded by exon 32. This case report inspired proof-of-concept studies to assess whether antisense-mediated exon skipping might have therapeutic merit for dysferlinopathies.
The exon skipping approach is most well known from Duchenne muscular dystrophy (DMD), an X-linked recessive disease caused by out-of-frame mutations (mainly large deletions) or nonsense mutations, which abolish production of functional dystrophin [11]. DMD is one the most severe neuromuscular disorders, with an early onset disease and very rapid progression. Like dysferlin, dystrophin is a modular protein [12]. Becker muscular dystrophy (BMD) is an allelic disorder with a later onset and a milder disease course than DMD. BMD patients generally carry large deletions that do not disrupt the reading frame and allow production of partially functional dystrophins that contain their crucial domains [13]. The exon skipping approach aims to modulate pre-mRNA splicing of DMD dystrophin transcripts such that the reading frame is restored and a BMD-like dystrophin can be produced [14]. The technique uses antisense oligonucleotides (AONs), small modified pieces of DNA or RNA that target a specific exon, generally one flanking an out-of-frame deletion. Hybridization to sequences within or near the exon hides it from the splicing machinery, thus bringing about the skipping of the targeted exon and restoration of the reading frame. This approach is mutation specific as depending on the size and location of the mutation, skipping of one or several specific exons is needed for reading frame restoration [15]. Eteplirsen, an exon 51 skipping AON, applicable to 13%–14% of DMD patients, has recently received accelerated approval by the Food and Drug Administration (USA) [16]; however, the European Medicines Agency (EMA) did not approve it [17].
For dysferlinopathies, the principle of exon skipping varies slightly from that for DMD, in that target exons do not flank the mutation, but carry the mutation [18,19]. When these exons are in frame, exon skipping would bypass the mutation without affecting the reading frame. When the mutated exon is not in frame, a combination of two or more exons will need to be skipped to bypass the mutation while maintaining the reading frame [18]. Like for DMD, the approach is mutation specific, depending on which exon is mutated. Thus far, few reports are available on dysferlin exon skipping in control and patient-derived cell cultures [18–22]. The focus has been primarily on exon 32 skipping, which was shown to increase expression of dysferlin and restore membrane healing deficits in vitro using wound healing and osmotic shock assays [20]. Furthermore, in cultured cells from a patient with an intronic mutation, which activated the inclusion of a pseudoexon in dysferlin transcripts, AON treatment resulted in the skipping of this pseudoexon and production of low levels of wild-type dysferlins [21]. Finally, AON-induced double exon skipping of exons 28 and 29 in cells derived from a patient with a missense mutation of exon 28 did not increase dysferlin levels, but did result in improved membrane sealing ability in treated cells [22].
We previously showed that it is feasible to induce exon skipping for a number of in-frame dysferlin exons in control myotubes (exons 19, 25, 30, 32, and 34) [18]. In this study, we aimed to study the therapeutic potential of the exon skipping approach further by assessing whether exon skipping can restore dysferlin production in patient-derived cell cultures carrying various mutations.
Materials and Methods
AON design
AONs used in this study are shown in Table 1. AONs were designed using previously identified guidelines for dystrophin exons [23]. Briefly, AONs of 18–22 nucleotides were selected based on the presence of predicted exonic splicing enhancer sites, a GC% of 40%–60%, and targeting partially open sequences in the predicted secondary structure of the exon. All AONs contained 2′-O-methyl RNA with a full-length phosphorothioate and were ordered from Eurogentec (Belgium). A noneffective AON targeting dystrophin exon 47 (h47AON2) was used as a negative control in all experiments [24].
Antisense Oligonucleotides Used in This Study
“−” no exon skipping detected, “+” up to 25% exon skipping, and “++” over 25% exon skipping, as detected by the Agilent bioanalyzer.
Cell culture and AON transfection
Immortalized and primary myoblasts (Table 2) were cultured in Skeletal Muscle Cell Growth Medium (PromoCell, Germany) supplemented with 10% fetal bovine serum (10439–024; Gibco, United Kingdom), 1.5% GlutaMAX (Gibco), and 0.5% gentamicin (10 mg/mL; Sigma-Aldrich, USA). For differentiation, cells were seeded on a collagen-coated surface, and at ∼70% confluence, the proliferation medium was replaced by Dulbecco's modified Eagle's medium (Gibco) supplemented with 5% horse serum (Gibco), 2% GlutaMAX, and 0.8% glucose (Sigma-Aldrich). AON transfections were done after 2 to 5 days of differentiation (based on the presence of differentiated myotubes) with Lipofectamine 2000 (Thermo Fisher Scientific) or polyethyleneimine (PEI) (EXGen500 in vitro transfection reagent; MBI Fermentas, United Kingdom) according to the manufacturer's instructions using 25–800 nM AONs. RNA was isolated with the TriPure Isolation Reagent (Sigma-Aldrich) 1 day later, as previously described [18]. For protein analysis, cells were lysed by applying 60 μL of 4 × Laemmli sample buffer (BIO-RAD) and scraping the cells 3, 5, and 8 days after transfection based on the report by Dominov et al. [21]. We generally did not observe cell toxicity after transfection, except when high (>500 nM) concentrations were used for AONs 8-1, 8-2, 9-1, 9-2, and 30-2.
Cell Lines Used
Reverse transcriptase-polymerase chain reaction analysis
Reverse transcriptase-polymerase chain reaction (RT-PCR) was performed as described previously (Aartsma et al., 2010). For cDNA synthesis, 500 ng of total RNA was used with DYSF gene-specific reverse primers (primers upon request). For control cells, single PCR was performed with 35 cycles using 0.5 μL of cDNA in a 20-μL reaction with primers flanking the targeted exons. For patient-derived cells, a nested PCR was performed for an additional 30 cycles when single PCR did not result in a visible fragment. PCR fragments were analyzed on 2% agarose gel. Exon skipping was confirmed by Sanger sequencing of fragments isolated from gel using the QIAquick Gel Extraction Kit (QIAgen, Germany). Exon skipping levels were semiquantitatively assessed with the high-sensitivity DNA kit (Agilent) using the Agilent 2100 Bioanalyzer, using manufacturer's instructions. Primer sequences can be find in Supplementary Table S1.
DNA analysis
DNA analysis was done to confirm the mutations in the DYSF gene. Genomic DNA (gDNA) was isolated with the Wizard® gDNA purification kit (Promega, USA), followed by PCR with intronic primers flanking the targeted exon under the same conditions used for RNA analysis. PCR fragments were cleaned up with the MinElute PCR Purification Kit (QIAgen) and analyzed by Sanger sequencing.
Western blot analysis
Cells were directly scraped with Laemmli buffer and lysates loaded on 3%–8% Criterion XT Tris-Acetate gels (BIO-RAD Laboratories, USA). The proteins were blotted on nitrocellulose membranes (BIO-RAD Laboratories) using the Trans-Blot Turbo transfer system (BIO-RAD Laboratories) and blocked with 5% milk powder (ELK, Campina, the Netherlands). The blots were stained overnight with NCL-Hamlet mouse-α-dysferlin (1:300; Leica Biosystems, United Kingdom), rabbit-α-USP-7 (1:1000; GeneTex, USA), and mouse-α-vinculin (1:1000; Sigma). For detection, the secondary antibody IRDye 800CW goat anti-mouse IgG (dilution 1:5000; LI-COR Biosciences, USA) was used to detect dysferlin and vinculin, while IRDye 680LT donkey anti-rabbit IgG (1:10,000; LI-COR) was used to detect USP-7 and the protein size marker, respectively, as described previously [16,25]. The blots were scanned using the Odyssey® CLx Infrared Imaging System (LI-COR Biosciences) and the band density of proteins was quantified using densitometric software (Odyssey, version 1.2; LI-COR Biosciences).
Results
Exon 30 skipping in control and patient-derived cells
Our initial focus was on exon 30 skipping because we had previously designed very efficient AONs targeting this exon (30-1 and 30-2) [18]. Furthermore, patient-derived cells with an exon 30 mutation were available in EuroBioBank. Before embarking on studies in patient cells, we first wanted to test whether exon 30 skipping could result in stable dysferlin protein. We therefore transfected human control myotubes (KM155) with AON 30-1 or 30-2 and isolated RNA 1 day after transfection and protein 3, 5, and 8 days after transfection. Exon 30 skipping was observed for all samples treated with AON 30-1 or 30-2, but not for untreated or control AON-treated samples (Fig. 1A). A dysferlin lacking the amino acids encoded by exon 30 would be predicted to be 7 kDa shorter. On western blotting, indeed, a faint extra band could be observed for 30-2-treated samples, but not for untreated or control AON-treated samples (Fig. 1B).
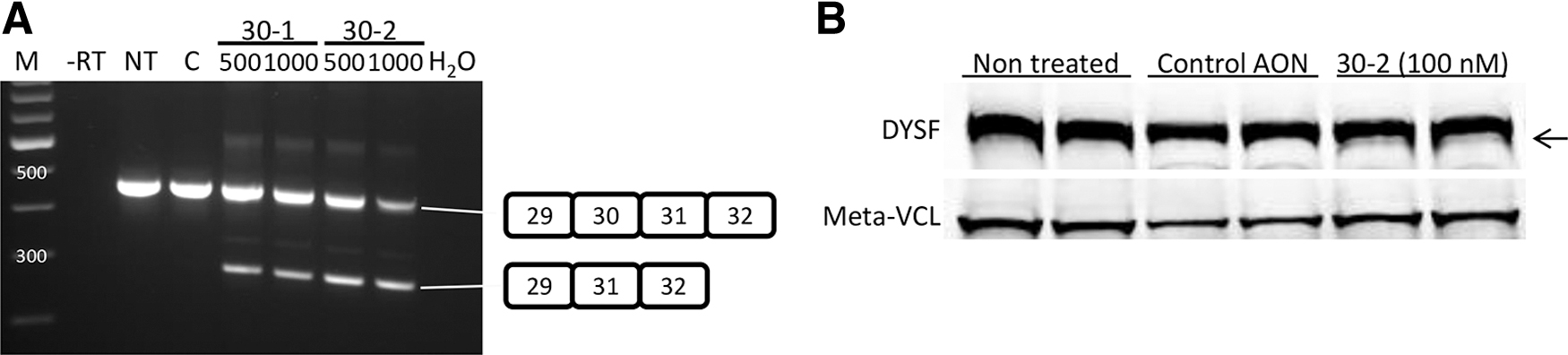
Exon 30 skipping in control myotubes.
We then initiated the culture of patient cell line 393, carrying an insertion mutation in exon 9 (c863dupA) and a variation of unknown significance in exon 30 (c3191_3196dup) (previously reported as patient 1 in Table 1, Walter et al.[26]). We confirmed the presence of the insertion in exon 30 by Sanger sequencing (Supplementary Fig. S1). Cells proliferated poorly and differentiation was also less efficient than for the control cell line, as has been reported previously for dysferlin-deficient cells [27]. Myotubes were transfected with 30-1 or 30-2, and exon 30 skipping was observed by RT-PCR analysis on RNA isolated 1 day post-transfection (Fig. 2A, B). Notably, 30-2 was less effective than 30-1, while in control cells, these AONs had similar efficiency. While the AONs did not target the mutation, it is possible that the presence of the mutation influenced AON efficiency. Protein was isolated 3–8 days after transfection for western blot analysis (Fig. 2C). Interestingly, a faint band could be observed in nontreated samples at the same height as control dysferlin. However, the intensity of this band did not increase upon 30-1 or 30-2 treatment, nor did an additional band appear.
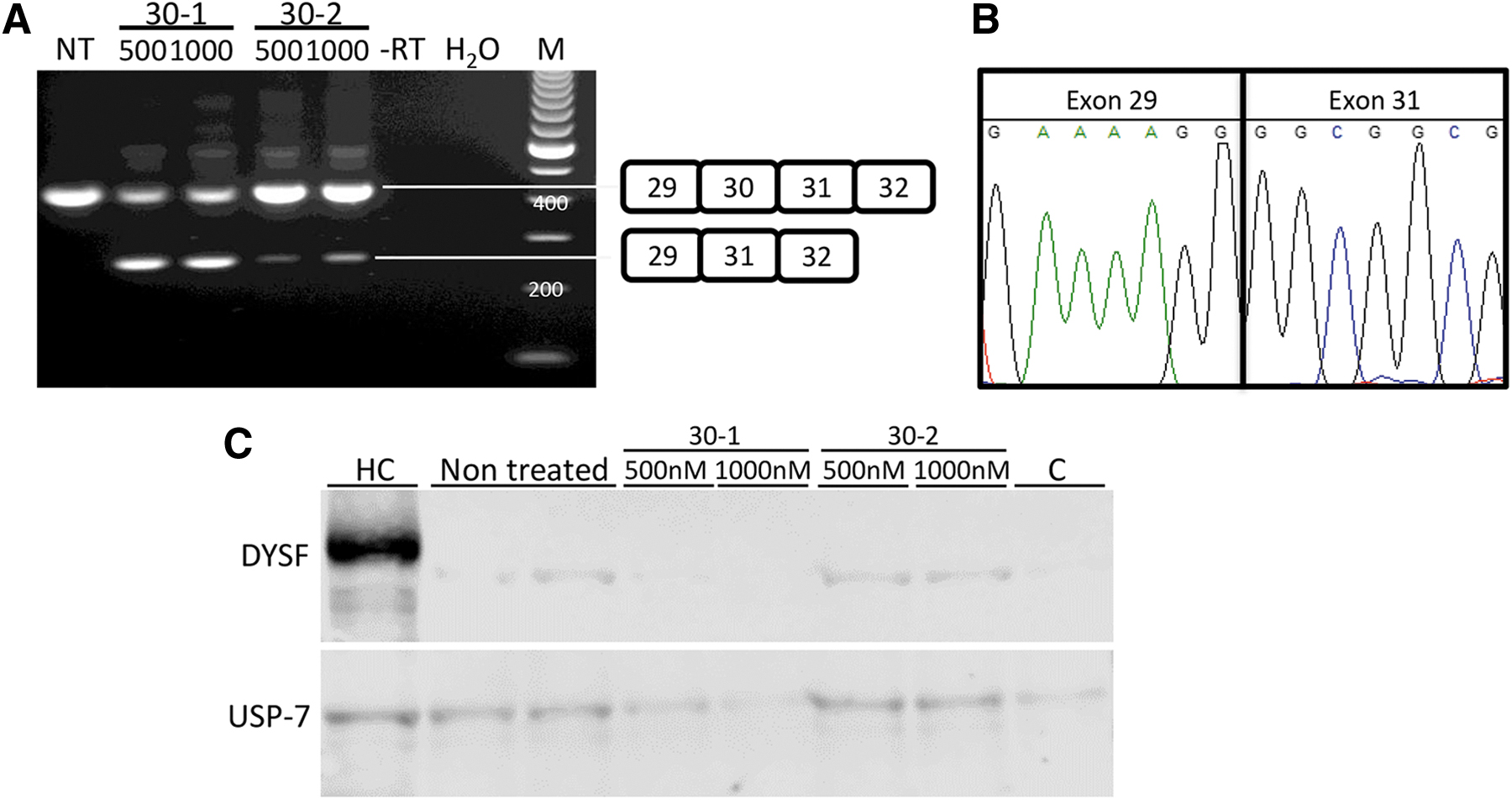
Exon 30 skipping with 500 and 1000 nm AON using PEI in 393-derived myotubes. Cell line 550 (HC)-derived myotubes were used as control.
Finding a second mutation for the 393 cell line
Since the 6-bp insertion in exon 30 is not anticipated to disrupt the reading frame, but rather cause the insertion of two amino acids, we wondered whether this mutation is truly disease causing. We discovered that while this insertion has been reported as a disease-causing mutation for a number of LGMD2B patients, it has in fact also been found in healthy individuals [28]. We thus concluded that the exon 30 insertion was not disease causing for patient 393 and embarked on identification of the true second mutation. RT-PCR analysis using primer pairs covering the complete dysferlin mRNA revealed a shorter band in the amplicon containing exons 24–32 (Fig. 3A). Sanger sequence analysis revealed that this was due to the skipping of exon 28 (Supplementary Fig. S1). PCR analysis on DNA isolated from the cell line revealed a T to C substitution in the splice donor of intron 28 (c3031 + 2T>C) (Fig. 3B). This substitution was present in the allele that contained the insertion in exon 30 (data not shown). Since exon 28 is an out-of-frame exon (106 bp), this mutation will lead to a frameshift and, as such, this can be considered the second disease-causing mutation for this patient. This finding has been communicated to the provider of the cell line. It turned out that they had independently identified the splice site mutation in intron 28 for this patient.
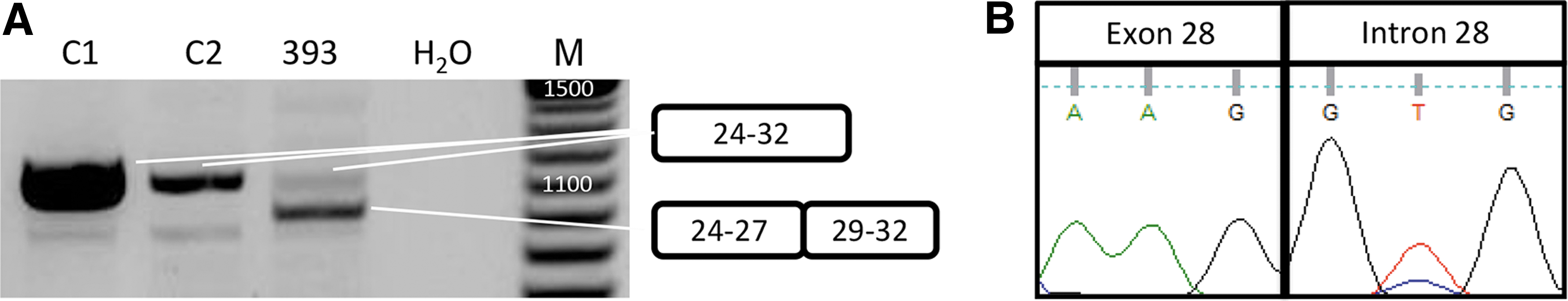
Identification of a second mutation for 393.
Exon 8, 9, 20, 29, and 34 skipping in control cell lines
We then made an inventory of available myoblast and fibroblast cell lines in various biobanks, prioritizing those available in-house or carrying mutations that could be corrected by skipping of a single exon encoding a (likely) redundant domain (2). We identified cells from patients with compound heterozygous mutations affecting exons 8, 9, 19, 20, 28, and 34 and one line from a patient with a homozygous mutation in exon 38. In theory, each mutation was considered eligible for exon skipping, except for the mutation in exon 19, since a splice site mutation causing the skipping of exon 19 has been reported as a disease-causing mutation previously [5,29]. Therefore, while exon 19 skipping is technically feasible, the resulting dysferlin is likely not functional. Exons 8, 9, 34, and 38 are in-frame exons encoding C2 domains and, as such, would be good candidates. The exon 28 splice site mutation identified for line 393 causes an out-of-frame skip. However, exon 29 skipping would restore the reading frame. Exons 28 and 29 encode predicted Dys-N-2 and Dysf-C-1 domains, and the exons 28 and 29 deleted dysferlin was recently shown to maintain membrane sealing ability [22]. The point mutation in exon 20 would require skipping of both exons 20 and 21 since exon 20 is out of frame. Exons 20 and 21 do not encode any predicted domain in the dysferlin protein.
Since no AONs had yet been identified for the skipping of exons 8, 9, 20, 21, 29, and 38, we designed AONs targeting these exons using previously described guidelines. For exon 34, AONs were available.
AONs were transfected in control myotubes at different concentrations and RNA was isolated 1 day after transfection. In each case, a nonspecific control AON was taken along as a negative control. RT-PCR revealed exon skipping of most of the targeted exons at various levels (Fig. 4). Very poor exon skipping could be observed for exon 8 and 21 AONs, while no exon skipping was observed for cells treated with exon 20 targeting AONs or the control AON. By contrast, exon 9, 29, 34, and 38 skipping was effective. Correct exon skipping was confirmed by Sanger sequencing (Supplementary Fig. S1).

Exon skipping of dysferlin exons 8, 9, 21, 29, and 38 in control myotubes.
Exon 9 and 34 skipping in patient-derived cells
Exon 9 and 34 skipping was most efficient. We therefore selected cell lines 107 (requiring exon 9 skipping) and 180 (requiring exon 34 skipping) to assess whether exon skipping could increase dysferlin levels in vitro. Cells were transfected with 200 nM of the respective AONs. RNA was isolated 1 day after transfection and skipping of the intended exons confirmed by RT-PCR (Fig. 5A). Protein was isolated 3 days later. Western blotting revealed the presence of dysferlin in untreated cells for both cell lines (Fig. 5B). No increases in dysferlin expression could be observed after treatment with the respective AONs or the control AON (repeated 9 times for exon 9 skipping and 13 times for exon 34 skipping).
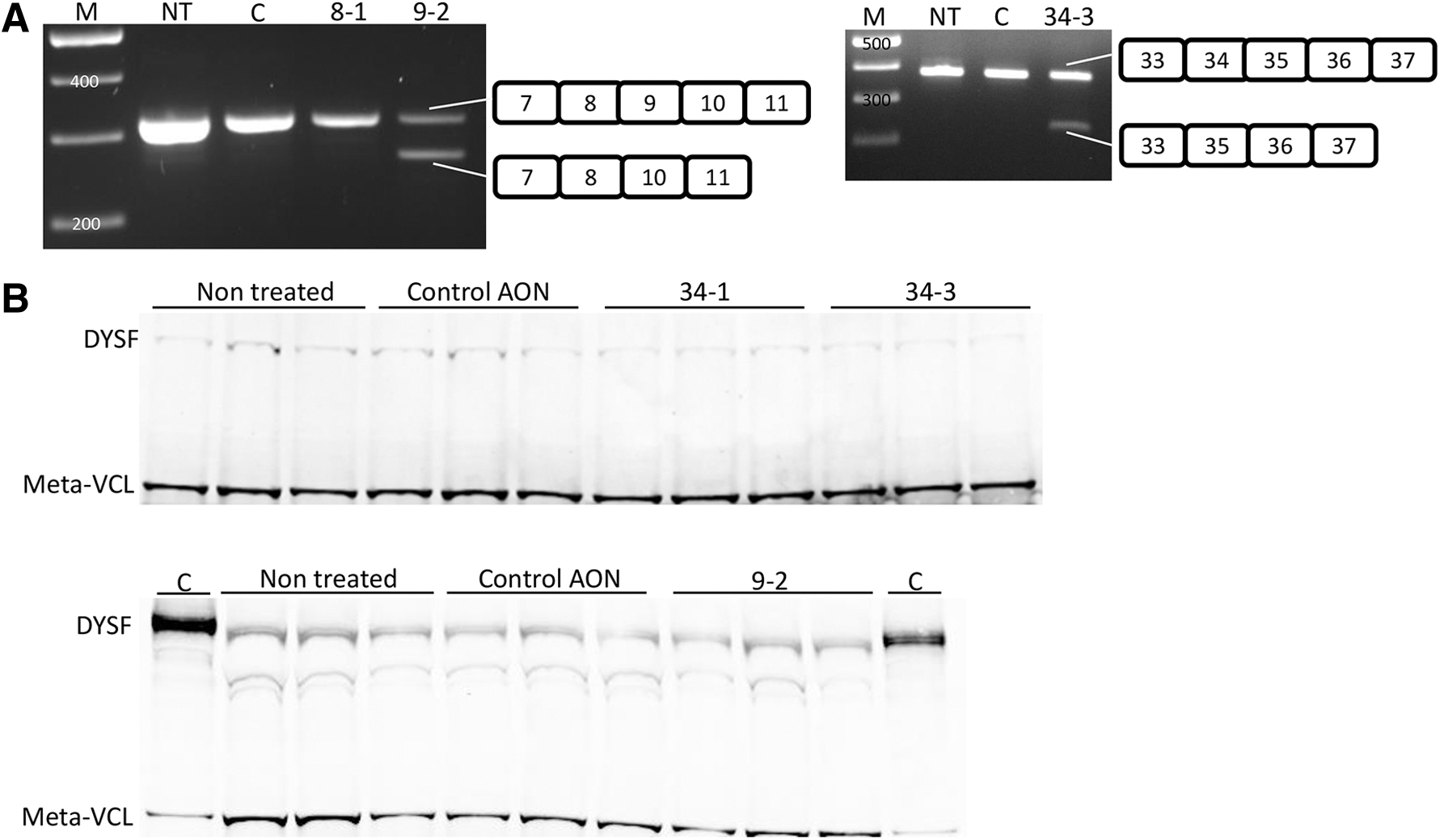
Exon 9 and 34 skipping in patient-derived myotubes.
Discussion
In this report, we show that it is feasible to induce skipping of dysferlin exons 8, 9, 21, 29, and 38 in control cells using AONs targeting exon-internal sequences. Combined with the exons that could already be skipped (exons 19, 24, 30, 32, and 34 [18]) and exon 28 [22]), it is now possible to skip 11 of the 55 dysferlin exons. Effective AONs to induce exon 20 skipping have thus far not been identified despite extensive efforts (7 AONs tested). However, when using various combinations of targeting AONs, exon 20 skipping could be induced (data not shown). This phenomenon has been reported before by us and others for dystrophin exon skipping [29,30] and is speculated to reflect the presence of two, strong, exonic splicing enhancer sequences in the target exon, where exon skipping requires the blocking of both.
We initially planned to test exon 30 skipping in a patient-derived myoblast cell line, but were unable to do so since the alleged mutation in this exon turned out to be a nondisease-causing variation. The alleged mutation would introduce 2 amino acids in exon 30 and, as such, would be unlikely to be disease causing. Indeed, this variation has been reported before also in unaffected individuals [28]. Further RNA analysis revealed exon 28 skipping caused by a mutation of the donor splice site of exon 28 in the allele that contained the exon 30 polymorphism. It is estimated that for ∼17% of dysferlinopathy patients, only one mutation has been identified [6]. It is possible that a number of these patients carry a mutation that affects splicing, for example, intronic changes that introduce the inclusion of a pseudoexon [21]. When possible, RNA analysis should therefore be performed for patients with a differential diagnosis of LGMD2B or Miyoshi myopathy, especially those for whom decreased dysferlin levels have been detected by protein analysis and/or those for whom a mutation can be found in only one allele.
A challenge of dysferlinopathies is that about half of the patients carry compound heterozygous mutations. For these patients, skipping a specific exon will only lead to dysferlin restoration for the transcripts produced by the allele with a mutation in the target exon. However, based on the fact that 10% of dysferlin is sufficient to generate a very mild phenotype, we speculate that bypassing the mutation only for transcripts arising from one allele may be enough, provided that the exon skipping is efficient enough to produce significant amounts of dysferlin. In fact, targeting both mutations for compound heterozygotes may be detrimental rather than productive. The cell line 107, for instance, was isolated from a patient who carries mutations in exons 8 and 9, both of which can be skipped. However, when AONs targeting both exons would be used, this would result in a mix of outcomes, some of which would be productive (exon 8 skipping in the exon 8 mutated transcript and exon 9 skipping in the exon 9 mutated transcript), some of which would not be productive (exon 9 skipping in the exon 8 mutated transcript, exon 8 skipping in the exon 9 mutated transcript, and no exon skipping), and for some, it is unknown whether the result would be productive (exon 8 and 9 skipping for both transcripts).
We tested both exon 9 and 34 skipping in patient-derived cells. For neither, however, were we able to confirm that this resulted in dysferlin restoration by western blotting. The analysis was impeded by the fact that for all cell lines tested (393, 107, and 180), low levels of dysferlin were present also before treatment. Despite efficient exon skipping levels, no increases in dysferlin expression could be observed for lines 107 and 180 after exon 9 and 34 skipping, respectively. We performed immunofluorescence analysis for line 107, but also here results were inconclusive and there was no clear increase in dysferlin-positive cells or fluorescent signals after AON treatment. We speculate that perhaps the skipped transcripts are sequestered and therefore not available for translation or that the produced dysferlin is unstable.
The presence of dysferlin in untreated patient cells fits with the report by Lee et al., where dysferlin levels were present before treatment and unaltered despite efficient exon skipping. The authors performed membrane sealing experiments showing that the dysferlin produced after exon skipping had increased membrane sealing functionality [22]. We were unable to perform membrane sealing experiments. As such, we do not know whether the resulting dysferlin proteins are functional. In fact, it is possible that the dysferlins encoded by transcripts lacking exon 9 or exon 34 are not functional. Splice site mutations have been reported as disease-causing mutations for in-frame exons 19, 25, and 49, and it has been suggested that exon 9, 24, 30, 34, 37, and 41 skipping might be pathogenic [29,31]. We have here confirmed that at least exon 30 skipping results in the expression of a stable protein since we could detect the protein by western blotting after transfection in control cells. Studies to assess localization of this internally deleted dysferlin are, however, not feasible since in control cells, the vast majority of proteins will be full-length proteins. As such, we currently cannot conclude whether dysferlin exon skipping has therapeutic potential beyond the previously reported exon 32 skipping and exon 28 and 29 double skipping [20,31]. Future studies using more efficient AONs, improved transfection methods, and/or transgenic animal models may reveal whether or not skipping additional exons will be beneficial.
As mentioned, dysferlinopathies are very rare and mutations are distributed over the transcripts. Thus, the groups to which skipping an individual exon will in theory apply are very small. This will make clinical development of personalized genetic approaches such as exon skipping very challenging for this disease [32].
Footnotes
Acknowledgments
The platform for immortalization of the Myology Institute in Paris is acknowledged for the control cell line. The authors also thank Muscle Tissue Culture Collection (MTCC) at Klinikum der Universität München for providing the samples.
Author Disclosure Statement
A.A.R. discloses being employed by LUMC, which has patents on exon skipping technology, some of which has been licensed to BioMarin and subsequently sublicensed to Sarepta. As coinventor of some of these patents, A.A.R. is entitled to a share of royalties. A.A.R. further discloses being ad hoc consultant for PTC Therapeutics, Sarepta Therapeutics, CRISPR Therapeutics, Summit PLC, Alpha Anomeric, BioMarin Pharmaceutical, Inc., Eisai, Astra Zeneca, Santhera, Audentes, Global Guidepoint, GLG consultancy, Grunenthal, Wave, and Bioclinica, having been a member of the Duchenne Network Steering Committee (BioMarin), and being a member of the scientific advisory boards of ProQR, Hybridize Therapeutics, Silence Therapeutics, Sarepta Therapeutics, and Philae Pharmaceuticals. Remuneration for these activities is paid to LUMC. LUMC also received speaker honoraria from PTC Therapeutics and BioMarin Pharmaceuticals and funding for contract research from Italfarmaco and Alpha Anomeric. All other authors have nothing to disclose.
Funding Information
The Muscle Tissue Culture Collection is part of the German network on muscular dystrophies (MD-NET) and the German network for mitochondrial disorders (mito-NET, 01GM1113A) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany). The Muscle Tissue Culture Collection is a partner of Eurobiobank (www.eurobiobank.org) and TREAT-NMD (![]() ). The work in this report was, in part, covered by funding from FP7 project Neuromics (grant no. 2012-305121).
). The work in this report was, in part, covered by funding from FP7 project Neuromics (grant no. 2012-305121).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.