
Abstract
While small interfering RNAs (siRNAs) are commonly used for laboratory studies, development of siRNA therapeutics has been slower than expected, due, in part, to a still limited understanding of the endocytosis and intracellular trafficking of siRNA-containing complexes. With the recent characterization of multiple clathrin-/caveolin-independent endocytic pathways, that is, those mediated by Graf1, Arf6, and flotillin, it has become clear that the endocytic mechanism influences subsequent intracellular processing of the internalized cargo. To explore siRNA delivery in light of these findings, we developed a novel assay that differentiates uptake by each of the endocytic pathways and can be used to determine whether endocytosis by a pathway leads to the initiation of RNA interference (RNAi). Using Lipofectamine 2000 (LF2K), we determined the endocytosis pathway leading to active silencing (whether by clathrin, caveolin, Arf6, Graf1, flotillin, or macropinocytosis) across multiple cell types (HeLa, H1299, HEK293, and HepG2). We showed that LF2K is internalized by Graf1-, Arf6-, or flotillin-mediated endocytosis for the initiation of RNAi, depending on cell type. In addition, we found that a portion of siRNA-containing complexes is internalized by pathways that do not lead to initiation of silencing. Inhibition of these pathways enhanced intracellular levels of siRNAs with concomitant enhancement of silencing.
Introduction
Small molecule and protein-based drugs, while critically important therapies, cannot treat all diseases [1]. In some cases, the drugs cannot access or interact with proteins that are causing the disease phenotype. As such, alternative treatment modalities must be developed to complement existing strategies. One potential alternative is small interfering RNA (siRNA) therapeutics, which are capable of specific inhibition of a wide range of intracellular, membrane, and extracellular proteins [2]. To function, siRNAs must be transported from the extracellular environment into the cytoplasms of the targeted eukaryotic cells. Once there, siRNAs act through RNA interference (RNAi) to degrade messenger RNAs (mRNAs) in a sequence-specific manner, thereby reducing target protein expression [3–7]. siRNA therapeutics are being developed as treatments for a variety of diseases, including cancers and infectious diseases, with one therapeutic approved for clinical use [8–10]. Despite the recent clinical success, development of siRNA therapeutics has been hindered by multiple technical challenges, including poor delivery efficiency [11–13]. One limitation to delivery is efficient endocytosis of delivered siRNAs to the cells of interest.
Until recently, cellular endocytic pathways were classified as macropinocytosis (MP), clathrin-mediated endocytosis (CME), caveolin-mediated endocytosis (CvME), or clathrin-/caveolin-independent endocytosis (CCIE) [14]. Researchers have since characterized three distinct types of CCIE, flotillin-mediated endocytosis (FME), Arf6-dependent endocytosis (ADE), and Graf1-mediated endocytosis (GME) [15–17]. The identification of these pathways has resulted in the reclassification of the uptake mechanisms of many species [18–20]. For instance, adeno-associated viruses and ∼50% of fluid-phase uptake, including uptake of dextrans, are now attributed to GME, although they were previously thought to occur through other pathways [16,21]. Likewise, cholera toxin B is taken up by FME, but was previously thought to enter cells by MP [15].
Currently, there is no consensus regarding the optimal endocytic pathway for active siRNA delivery, as multiple endocytic pathways have been found to result in successful delivery of siRNAs and initiation of silencing. It is difficult to generalize which pathways are optimal as most studies are limited to a single cell type or did not distinguish among FME, ADE, and GME. However, it has also recently become evident that the endocytic mechanism influences the molecular composition of the endosomes, their intracellular trafficking, and the processing of their cargo [22,23]. Thus, we hypothesized that the mechanism used by cells to endocytose siRNA-containing complexes could significantly impact the ability of the siRNAs to initiate RNAi.
In this study, we used chemical inhibitors and endocytic protein overexpression to investigate the endocytic pathways used to internalize and process siRNA-containing complexes in four cell lines. Our results show that, while the complexes are internalized through multiple endocytic pathways, active delivery occurs primarily through a single pathway that varies according to cell type. The results suggest that both cell specificity and siRNA delivery efficiency can be enhanced by designing delivery vehicles to favor the preferred endocytic pathway.
Materials and Methods
Materials
See Supplementary Table S1 for a detailed list of reagents and solutions.
Cell lines
EGFP-expressing H1299 and HeLa cells were generously provided by Dr. Jørgen Kjems and Dr. Manfred Gossen, respectively [24,25]. HepG2 and HEK293 cells constitutively expressing EGFP (HepG2-EGFP and HEK293-EGFP) were generated using the methods outlined in Gossen and colleagues [25]. Briefly, cells were seeded in six-well plates and transfected 24 h postseeding with 4 μg pEGFP and 10 μL Lipofectamine 2000 (LF2K). Three days post-transfection, cells were sorted and replated according to their EGFP expression using a flow cytometer. This process was repeated at 7 and 14 days post-transfection. The average EGFP expression of the final population was analyzed over several cell cycles and found to be stable. All cell lines were maintained in antibiotic-free Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were incubated at 37°C in 5% CO2, at 100% relative humidity, and subcultured every 4–5 days by trypsinization.
EGFP silencing
EGFP-expressing cells were seeded in 24-well plates at 200,000 cells/well (400,000 cells/well for HepG2 cells) in 500 μL of antibiotic-free DMEM/FBS. Cells were treated 24 h postseeding with 100 μL of transfection solution containing Opti-MEM, siRNA, and LF2K, yielding final concentrations of 100 nM siRNA and 2.3 μg/mL LF2K. Cells were washed 4 h post-transfection with DMEM/FBS and incubated in Dulbecco's phosphate-buffered saline (DPBS; +Mg/Ca) containing 20 μg/mL heparin sulfate for 5 min to remove any extracellular siRNAs. The heparin sulfate solution was subsequently removed and replaced with DMEM/FBS. At 24 h post-transfection, cells were trypsinized, fixed in 2% paraformaldehyde [v/v in DPBS (-Mg/Ca)], and stored in DPBS (-Mg/Ca) at 4°C until analysis (typically less than 3 days; results were stable 24 days postfixation). Cells were analyzed by using a Becton Dickinson Influx Flow Cytometer to detect both EGFP (488/530) and Dy547 tagged siRNA (557/574) signal within each event. Samples were gated to include 10,000 events/sample, where only cells with a positive siRNA signal were counted as events. EGFP fluorescence was measured using an excitation of 488 nm with a multiline Argon laser. Dy547-tagged siRNA fluorescence was excited at 552 nm by a HeNe laser. Geometric mean was used to calculate fluorescence intensity values among samples. Propagation of error was used to minimize type 1 errors. Incubations were conducted at 37°C, 5% CO2, and 100% humidity. Cell morphology and EGFP expression as a measure of cytotoxicity were assessed by microscopy and were not significant in any of the treatments.
Endocytic inhibitors
Endocytic inhibitors were used for 5 h at concentrations based on the literature and our own toxicity and dose–response experiments (Table 1, Supplementary Table S2, and Supplementary Fig. S1). The specificity (or lack thereof) of the inhibitors was assessed from the literature yielding a logic matrix that allows for differentiation of the function of different endocytic pathways through comparison of the effects of multiple inhibitors (Supplementary Table S3).
Chemical Inhibitors of Endocytic Proteins
For general information about inhibitors, see Ivanov [47].
MβCD, methyl-β-cyclodextrin.
Inhibition experiments
EGFP-expressing cells were seeded in 24-well plates at 200,000 cells/well (400,000 cells/well for HepG2 cells) in 500 μL of antibiotic-free DMEM/FBS. After 23 h, cells were washed with DMEM and incubated for 1 h in DMEM containing inhibitors (Table 1). Cells were then transfected with siRNA as above. Cells were washed 4 h post-transfection with antibiotic-free DMEM/FBS and incubated in heparin sulfate solution for 5 min to remove extracellular siRNAs. The heparin sulfate solution was subsequently removed and replaced with antibiotic-free DMEM/FBS. At 24 h post-transfection, cells were trypsinized, fixed using a 2% paraformaldehyde solution, and stored in DPBS (-Mg/Ca) at 4°C until analysis. All incubations were conducted at 37°C, 5% CO2, and 100% humidity. Cells were then analyzed by flow cytometry and microscopy (Supplementary Figs. S2 and S3) as above.
Co-culture inhibitor experiments
HeLa, H1299, HEK293, and HepG2 cell lines, only one expressing EGFP, were mixed and seeded into 24-well plates at a density of 50,000 cells/well for HeLa, H1299, and HEK293 and 100,000 cells/well for HepG2 (total cell concentration of 250,000 cells/well) in 500 μL of DMEM/FBS. Cells were treated with siRNAs, fixed, and analyzed by flow cytometry as above.
Endocytic protein overexpression experiments
Cells were seeded in 24-well plates at a density of 150,000 cells/well (300,000 cells/well for HepG2 cells) in 500 μL of antibiotic-free DMEM/FBS. After 24 h, cells were transfected with a 100 μL transfection solution containing Opti-MEM, Lipofectamine 3000 (LF3K), and one of the following plasmids: pd2EGFP-N1 (EGFP; control), wt dynamin 2 pEGFP (Dynamin), EGFP-Actin-7 (Actin), GFP-alpha-adaptin (AP2) (kindly provided by Rappoport et al. [26]), GFP-clathrin (Clathrin) (kindly provided by Keen and colleagues [27]), Cav1-GFP (Caveolin), pFlot-1-GFP-N1 (Flot 1) (kindly provided by Tikkanen and colleagues [28]), pFlot-2-GFP-N1 (Flot 2) (kindly provided by Tikkanen and colleagues [28]), pDEST47-ARF6-GFP (Arf6), and pEGFP-C3-GRAF1 (Graf1) (kindly provided by Lundmark et al. [17]). Concentrations (after addition to the growth media) were optimized for both toxicity and expression level: HeLa and H1299 (150 ng plasmid, 0.55 μg LF3K), HEK293 (600 ng plasmid, 2.2 μg LF3K), and HepG2 (800 ng plasmid, 4.4 μg LF3K). Cells were washed 6 h after plasmid transfection with antibiotic-free DMEM/FBS. Twenty-four hours after plasmid transfection, cells were transfected with siRNAs, as above. Cells were then fixed and analyzed by flow cytometry and microscopy (Supplementary Fig. S4) as above.
Statistics
Statistical analyses were performed using one-way analysis of variance, followed by Tukey's HSD post hoc analysis. P values for all comparisons are provided in Supplementary Table S4.
Results
Silencing efficiency in different cell lines
To assess the role of endocytosis in siRNA accumulation and EGFP silencing, we tested the ability of LF2K to deliver siRNAs and achieve active silencing in four common human cell lines stably expressing EGFP: H1299 (lung), HeLa (cervical), HEK293 (kidney), and HepG2 (liver) (Fig. 1). At 24 h post-transfection, EGFP silencing and siRNA accumulation were measured in all cell lines. Silencing was greatest in H1299 cells (Fig. 1A), yet levels of intracellular siRNAs were highest in HEK293 cells (Fig. 1B), and the most efficient use of siRNAs (silencing/siRNA accumulation) was seen in HeLa cells (Fig. 1C). These differences suggested that the internalization and processing of LF2K-siRNA complexes differ among cell types, possibly due to the predominance of different endocytic pathways across the different cell types.
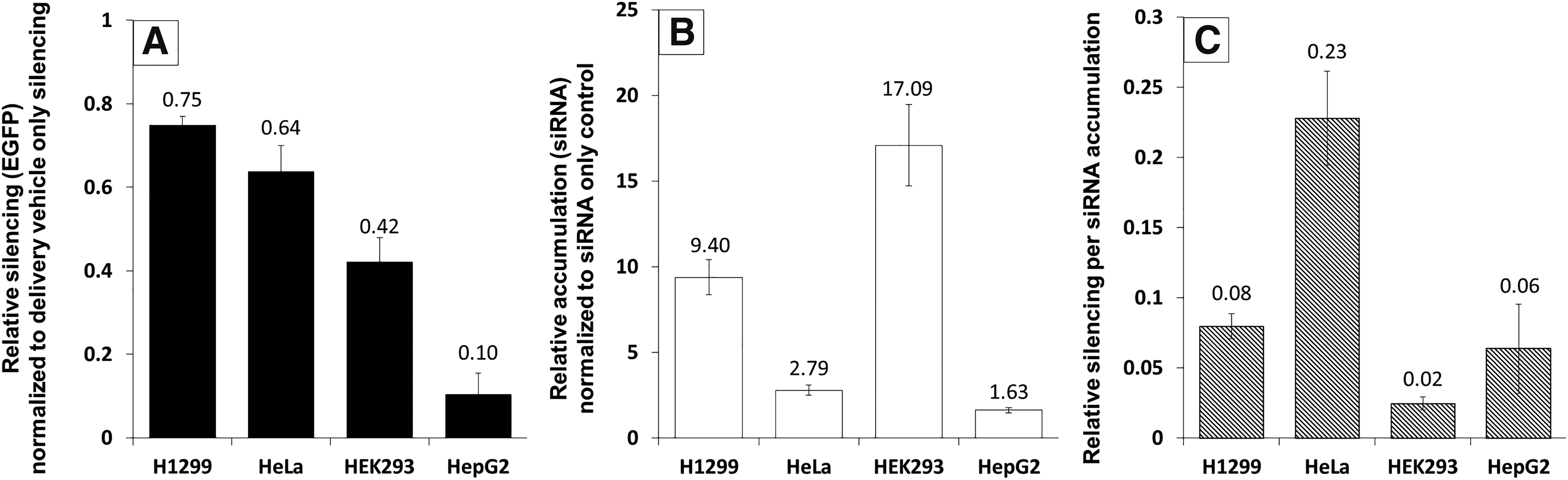
EGFP silencing and siRNA accumulation in H1299, HeLa, HEK293, and HepG2 cells. Cells were transfected with 100 nM fluorescently labeled siRNA and 2.3 μg/mL LF2K and assayed 24 h post-transfection using flow cytometry (10,000 events).
Inhibition of siRNA accumulation and silencing
It is known that drug complexes are taken into cells through multiple endocytic pathways. However, in most circumstances, it is unclear whether the mechanism of uptake influences downstream function of the complexes. To differentiate among the types of endocytosis, we used a minimal set of chemical inhibitors, which, when evaluated collectively, results in unique patterns of inhibition for each endocytic mechanism. Using data from the literature, a logic matrix was constructed for each inhibitor and its effect on each type of endocytosis (Supplementary Table S3). Using this logic matrix, we were able to identify the type of endocytosis used by LF2K for active siRNA delivery across each of the cell lines tested. By measuring the effect of inhibitors on both intracellular levels of siRNAs and EGFP silencing, we were also able to classify the endocytic pathways according to their role in facilitating siRNA function. Results were normalized against siRNA accumulation and silencing in the absence of inhibitor, allowing the relative position of a data point to indicate the degree to which an inhibitor affected siRNA accumulation and silencing (Fig. 2, also see Supplementary Equations in Supplementary Data).

Influence of endocytic inhibitors on EGFP silencing and siRNA accumulation. EGFP-expressing cells
In comparing the effects of the different inhibitors among the four cell lines, the strongest, most consistent inhibition of silencing (and siRNA accumulation) was from methyl-β-cyclodextrin (MβCD) (Fig. 2, vertical lines). This is not unexpected, as MβCD inhibits multiple endocytic pathways (Supplementary Table S3). MβCD, a cyclic oligomer of glucopyranoside, forms soluble inclusion complexes with cholesterol in the cell membrane, principally destabilizing lipid rafts [29]. Inhibition of EGFP silencing by MβCD demonstrates that endocytosis of LF2K-siRNA complexes by lipid raft-dependent pathways is critical for the initiation of RNAi in each of these cell types. Because EGFP silencing was inhibited by MβCD, but not filipin (Fig. 2, black with white dots), the critical pathways in these cells involve one or more of the following: FME, ADE, and GME.
Cytochalasin D significantly inhibited siRNA accumulation and EGFP silencing in all, but H1299, cells (Fig. 2B–D vs. Fig. 2A, horizontal lines). Cytochalasin D, a mycotoxin that binds to F-actin and blocks its polymerization, prevents the formation of endocytic vesicles as they bud from the plasma membrane [30]. FME, however, forms endocytic vesicles through actin-independent tubular invaginations and is unaffected by cytochalasin D [31,32]. Thus, we concluded that, in H1299 cells, FME of LF2K-siRNA complexes results in initiation of RNAi.
Dynasore also reduced EGFP silencing, but only in HEK293 and H1299 cells (Fig. 2A, B, gray). Dynasore, a noncompetitive inhibitor of dynamin, prevents endocytic vesicle fission from the cell membrane [33]. Among the lipid raft-dependent endocytic pathways, only ADE is considered dynamin independent [34]. Because EGFP silencing in HeLa and HepG2 cells was inhibited by MβCD and cytochalasin D, but not dynasore, we concluded that RNAi is initiated following ADE of LF2K-siRNA complexes in these cell lines.
Amiloride was the only other inhibitor to reduce EGFP silencing, but only in HEK293 cells (Fig. 2B, black). Amiloride, a derivative of a guanidinium-containing pyrazine, increases submembranous pH by inhibiting Na+/H+ exchangers [35]. Because EGFP silencing in HEK293 cells was inhibited by MβCD, dynasore, cytochalasin D, and amiloride, we concluded that GME is the principal RNAi-initiating pathway in HEK293 cells.
While inhibition of RNAi-initiating pathways is evident from reductions in EGFP silencing, inhibition of other pathways may also alter siRNA accumulation without a concomitant decrease in silencing. In H1299 cells, chlorpromazine significantly reduced siRNA accumulation without affecting EGFP silencing (Fig. 2A, white with black dots). Chlorpromazine, which translocates clathrin and AP2 from the plasma membrane to intracellular vesicles, inhibits the formation of clathrin-coated pits used in CME [36]. Therefore, we concluded that, in H1299 cells, CME internalizes siRNAs, but does not allow them to initiate silencing. In HeLa and HepG2 cells, chlorpromazine enhanced siRNA accumulation and EGFP silencing (Fig. 2C, D, white with black dots), suggesting inhibition of CME in these cells results in additional siRNAs entering ADE and initiating RNAi. However, the inhibitor data do not allow us to determine whether CME is also capable of internalizing siRNAs or if the enhancement of ADE results from an intracellular connection between ADE and CME. In HEK293 cells, amiloride inhibited uptake through GME, reducing silencing. The data show that this also resulted in additional siRNAs accumulating through an uninhibited pathway. As multiple endocytic pathways are unaffected by amiloride, these data alone were insufficient to identify the pathway(s) responsible for the enhanced siRNA accumulation.
Overexpression of endocytic proteins
To validate the findings from our inhibitor experiments and make additional distinctions between pathways, we overexpressed individual endocytic proteins and measured the effects on siRNA accumulation (Fig. 3). GFP-labeled proteins were used so that localization of the overexpressed protein could be confirmed to match that of endogenous protein (Supplementary Fig. S4), and to ensure that siRNA accumulation was only measured for cells overexpressing the protein. It is critical to note that, as in the inhibitor experiments, the effects of protein overexpression are cell specific (compare Fig. 3A–D).
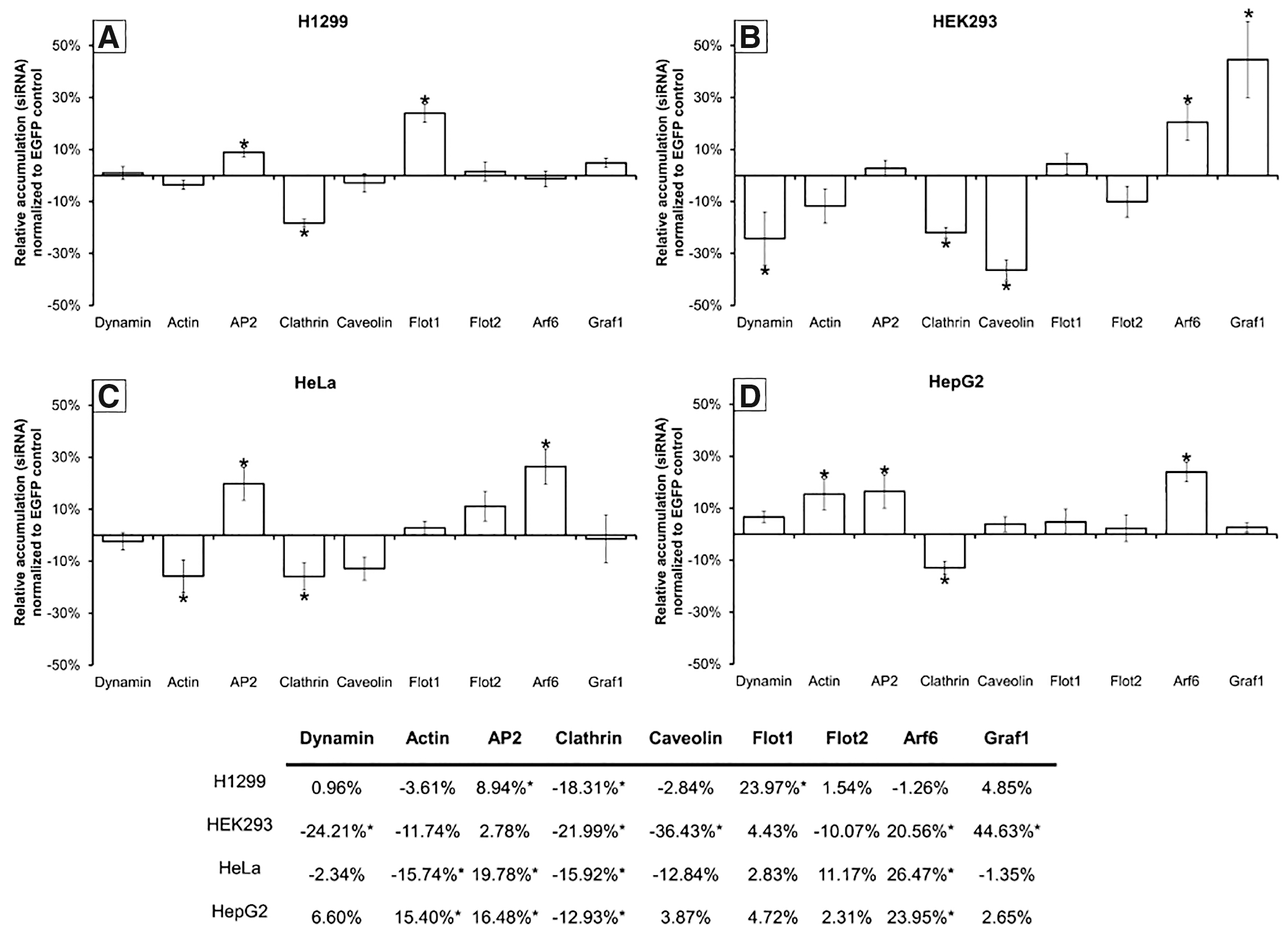
Influence of endocytic protein overexpression on the intracellular accumulation of siRNAs. Shown is the relative fluorescence of complexes containing fluorescently labeled siRNAs delivered to cells transiently expressing EGFP-labeled endocytic proteins
In H1299 cells, we found that siRNA accumulation was enhanced by overexpression of flotillin-1 and AP2, although reduced by clathrin overexpression (Fig. 3A). These findings indicate that both FME and CME are capable of internalizing siRNA, supporting the findings from our inhibitor data.
siRNA accumulation in HEK293 cells was enhanced by the overexpression of Graf1 and Arf6, although reduced by dynamin, clathrin, and caveolin overexpression (Fig. 3B). This would suggest that both GME and ADE are capable of internalizing siRNA-LF2K complexes. This supports our inhibitor results for GME in these cells. It also demonstrates that ADE can internalize siRNAs, although without leading to RNAi, and is likely responsible for the enhanced siRNA accumulation that occurred in the presence of amiloride (Fig. 2B). Similarly, our results suggest that CME, CvME, and GME share common regulatory elements, where overexpression of clathrin or caveolin dilutes the availability of these common elements for GME, resulting in reduced siRNA accumulation.
In HeLa and HepG2 cells, the accumulation of siRNAs was enhanced by overexpression of Arf6 and AP2, but reduced by overexpression of clathrin (Fig. 3C, D). These findings confirm that internalization of siRNA-LF2K complexes occurs through both ADE and CME, as in our inhibitor data. It is interesting that the cell lines show different responses to the overexpression of actin (Fig. 3C, D). This difference may partially explain why siRNA-LF2K complexes accumulate to a lesser degree and are considerably less efficient at initiating RNAi in HepG2 cells (Fig. 1C), although a direct mechanistic link is not currently known (see Discussion section).
Targeted inhibition in a co-cultured population
Having demonstrated that the pathways that are important for internalizing siRNAs and initiating RNAi vary by cell type, we theorized that inhibitors could be employed in a mixed cell population to enhance cell-specific delivery by reducing uptake by untargeted cell types. To test this, we repeated our inhibitor assay using a co-culture consisting of H1299, HEK293, HeLa, and HepG2 cells and assessed the effect of inhibitors on siRNA accumulation and EGFP silencing (Fig. 4). In general, the effects of the inhibitors in co-culture were the same as the effects on monocultures (Fig. 4, compare O and Δ). Three cases deviated from the monoculture results, treatment with cytochalasin D and chlorpromazine in H1299 cells (Fig. 4A, horizontal lines, white with black dots) and dynasore in HeLa cells (Fig. 4C, gray). For two of these cases, endocytosis of siRNA complexes by a specific cell type was enhanced by inhibition of endocytosis by other cell types.

Influence of endocytic inhibitors on EGFP silencing and siRNA accumulation in co-cultured and monocultured populations. Co-cultured populations consisted of H1299, HEK293, HeLa, and HepG2 cells. EGFP-expressing cells
Discussion
Using inhibition and overexpression of endocytic proteins, we showed that LF2K-siRNA complexes are internalized through multiple endocytic pathways. Moreover, the pathways used for endocytosis of LF2K-siRNA complexes were found to vary across cell types. The functional roles of these pathways were further characterized according to whether they facilitated LF2K-mediated RNAi. We also demonstrated that understanding the endocytic pathways of cells allowed targeting of specific cells in a mixed population and a resulting enhancement of siRNA accumulation and RNAi in the targeted cell populations.
We recognize that LF2K is not an option for future clinical applications and that delivery vehicle development has progressed since LF2K first became available. Nonetheless, we chose LF2K for these studies for two principal reasons. First, we have considerable prior experience using this vehicle [37–39]. Second, there is extensive prior literature on the use of this vehicle [40,41], allowing our results to be compared to the extant literature. We are not suggesting that the pathways used by LF2K are those that will be preferred by other vehicles. Rather, as our results show, the same vehicle works differently depending on the cell type, and uptake alone is not sufficient to achieve activity. These lessons can be applied to the development of any vehicle.
Previous studies regarding the cellular uptake of lipoplexes have reported that internalization occurs by CME or through direct fusion with the plasma membrane [42–45]. The differences in our conclusions relative to these prior studies may be a result of differences in the concentrations of inhibitors used, the presence of serum in the treatment media, wash procedures, or inhibitor exposure time. It may also be that the inhibitors chosen for this study, and an improved understanding of their impacts on cell function, allowed us to identify endocytic pathways with more clarity than was possible previously. We and others have shown that transfection at low temperature (4°C) reduces silencing [39,42], supporting our current conclusions that the best pathways for endocytosis of siRNA-containing complexes in the cell types tested are energy dependent and therefore do not include direct membrane fusion.
Chemical inhibitors, siRNAs, and protein overexpression are commonly used to characterize the function of endocytic pathways [20]. Our inhibitor logic matrix was derived from the current understanding of the proteins targeted by the inhibitors and their associations with each endocytic pathway, including any known side effect at the concentrations used in our experiments (Supplementary Table S3). We chose to use inhibitors, as they work more quickly than siRNAs and overexpression, and result in a shorter-term reversible disruption of native cell function. However, among the many chemical inhibitors used to evaluate endocytosis, none possesses absolute specificity for a single endocytic pathway [46]. In many cases, the molecular target of an inhibitor is utilized by multiple endocytic pathways. In addition, experimental conditions (high concentrations, prolonged incubation, and serum protein interactions) can cause unintended side effects [47]. For example, MβCD, which inhibits lipid raft-dependent endocytosis, can also inhibit CME when used at concentrations >10 mM [48]. Fluorescent endocytic markers are generally used to determine the effective concentration of an inhibitor. To date, however, none has been established that is specific for GME, ADE, or FME, and those traditionally associated with CvME (albumin) [49,50] and MP (dextran) [17] have been shown to be endocytosed through multiple pathways.
It is still unclear what factors impact whether pathways are used for endocytosis of siRNA-containing complexes or which pathways lead to initiation of RNAi. Intracellular trafficking of endocytic vesicles varies across cell type and disease state [51]. Many of these variations are observed in relationship to processing through the early endosome (EE), a common node among intracellular trafficking pathways. In HeLa cells, the time for cargo to reach the EE was 5–10 min by CME and 30–60 min for ADE [16]. FME is capable of retrograde transport directly to the Golgi, bypassing the EE [52]. ADE and GME have been shown to form intermediate endosomal compartments capable of sorting cargo before joining the EE [16,53,54]. These differences alone could explain the differences in siRNA accumulation and silencing across cell types. In addition, the pH and composition of the endosomal vesicles differ among endocytic pathways [55,56], which could alter endosomal escape, depending on the mechanism (eg, formation of membrane pores, pH buffering, or membrane fusion) [56]. Thus, differences in the endosomal release kinetics for each endocytic pathway, in addition to uptake, may result in the differences in siRNA activity we observed among the different endocytic pathways and cell types. Differences in release kinetics may also explain why the active endocytic pathway for uptake of drugs and other molecules differs depending on the cell type [42,57–60].
By measuring both the intracellular accumulation of siRNA and its functional activity in silencing EGFP, we identified multiple endocytic pathways used to internalize siRNA-LF2K complexes. In three cases, we observed a significant increase in siRNA accumulation (see Fig. 2B, black and Fig. 2C, D, white with black dots). In each case, we were able to identify a regulatory protein common to both the inhibited and enhanced pathway (Cdc42 for amiloride and AP2 for chlorpromazine), which was also directly affected by the inhibitor. Given the duration of incubation with inhibitor, it is unlikely that the increase in endocytic activity is caused by increased protein levels. It is more likely a reallocation of cellular resources. AP2, which regulates CME, is in turn regulated by Arf6. Sequestration of AP2 to intracellular compartments by chlorpromazine would, in theory, increase the availability of Arf6 for ADE. In this way, the relative activities of endocytic pathways are affected by competition for common resources. Indeed, if this is the case, the relative expression of endocytic and regulatory proteins in a cell may control the relative activities of the respective endocytosis pathways.
We concluded that FME facilitates LF2K-mediated RNAi in H1299 cells. In FME, flotillin-1 and flotillin-2 co-assemble into plasma membrane microdomains in lipid rafts and are internalized after phosphorylation by FYN [61]. Previously, FME has been implicated in the uptake of CD59 [15], cholera toxin B [15], silica nanoparticles [62], and cationic polyplexes [63]. The role of dynamin in this process, however, is still undefined and possibly dependent on cell type or cargo [64]. Based on our inhibitor data with dynasore, we concluded that FME is dynamin dependent in H1299 cells. Interestingly, the progression of malignancy in non-small cell lung cancers, like H1299s, is characterized by increased expression of flotillin-2, and decreased expression of flotillin-1 and caveolin-1 [65]. This aligns with our findings where siRNA accumulation was unaffected by overexpression of flotillin-2, but enhanced by the overexpression of flotillin-1. Expression profiles of the mRNAs for the flotillins and caveolin-1 correlate across tissue samples, with the highest expression levels in heart, lung, and skeletal muscle tissue [66]. Using gene expression data, we found that ETS1, a transcription factor for both flotillins and caveolin-1, was 9.6x higher in H1299 cells than HeLa, HEK293, and HepG2 cells (Supplementary Table S5). This suggests that elevated expression of the flotillins, caveolin-1, or ETS-1 may facilitate uptake by FME and initiation of RNAi.
In HEK293 cells, we concluded that the cells use GME to initiate RNAi. Since its discovery, GME has been implicated in the uptake of GPI-linked proteins [17], adeno-associated virus [21], and dextran [67]. It was also identified as a major source of uptake of extracellular fluid [17]. In GME, Graf1 and dynamin form a stable complex that regulates the scission and stability of the tubulovesicular structures [17]. Interestingly, Graf1 in this complex has a higher affinity for dynamin-1 (DNM1), thought to be exclusive to neurons, than dynamin-2 (DNM2), which has ubiquitous expression [17,68]. Comparing gene expression data for Graf1, DNM1, and DNM2 among the four cell lines, we found that similar expression levels of DNM1, DNM2, and Graf1 only occurred in HEK293 cells (Supplementary Table S5). It is possible then that the relative expression levels of DNM1, DNM2, and GRAF1 determine the prominence of GME in a given cell type. In addition, mRNA expression levels of proteins associated with GME (Graf1, Cdc42, and Arf1) were significantly higher in HEK293 cells relative to the other cell lines tested (Supplementary Table S5).
ADE is regulated by the GTP cycle of Arf6 [16]. Internalization through ADE leads to the formation of Arf6-containing endosomes that are either recycled to the plasma membrane or trafficked to the EE, a process dependent upon the hydrolysis of Arf6-GTP [69]. ADE has currently been suggested as the route of internalization of Tac [53], major histocompatibility complex class I proteins (MHCI) [70], β-integrin [71], and the herpes simplex virus [72]. We found that both HeLa and HepG2 use ADE to initiate RNAi, although with different efficiency. In ADE, localization and phosphorylation of Arf6 are dependent upon actin polymerization [16]. Overexpression of actin in HeLa cells reduced siRNA accumulation, whereas in HepG2 cells, actin overexpression enhanced siRNA accumulation. Basal HeLa cell expression of actin mRNA is 2.3-fold higher than in HepG2 cells (Supplementary Table S5). Given the different responses of the cell types to actin overexpression, it may be that there is an optimal amount of actin to support ADE, with too much or too little being inhibitory.
We also showed that endocytic inhibitors could be used in a co-cultured population of cells to enhance silencing in multiple cell types or achieve preferential uptake in a given cell type (Fig. 4). This was principally observed through treatment with chlorpromazine in H1299, HeLa, and HepG2 cells (Fig. 4, white with black dots), cytochalasin D in H1299 cells (Fig. 4A vs. Fig. 4B–D, horizontal lines), and dynasore in HeLa cells (Fig. 4C vs. Fig. 4A, B, and D, gray). Given our results, we believe that controlling the design of siRNA delivery vehicles and accounting for the variability in endocytic pathways when delivering siRNAs could allow improved cell specificity in vivo, thereby enhancing the overall delivery efficiency and efficacy of siRNA-based therapeutics.
Although the specific pathways utilized by LF2K are, almost certainly, not ubiquitous among delivery systems, our findings demonstrate that (1) uptake alone is not sufficient to achieve silencing and (2) the role of endocytosis in siRNA therapeutics warrants additional study. Overall, these findings also support a growing body of evidence that the endocytic pathway used for internalization is dependent on cell type, in addition to the characteristics of the cargo. For the field of siRNA therapeutics, these findings suggest that delivery vehicles should be designed to utilize specific endocytic pathways when targeting a particular cell type. By simultaneously enhancing uptake through pathways that initiate RNAi and avoiding uptake through pathways that do not, the efficacy and specificity of siRNA-based therapeutics could be markedly enhanced.
Footnotes
Acknowledgments
We would like to thank the members of the Cellular and Biomolecular Laboratory for their advice and support, especially McKenna Coskie, Hannah Cavagnetto, Harrison Lawson, and Chauncey Splichal, for their assistance with this work. We thank Dr. Melinda Frame and the Michigan State University Center for Advanced Microscopy for support of our confocal microscopy experiments. We would also like to thank Dr. Louis King for support of our flow cytometry experiments.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Financial support for this work was provided, in part, by Michigan State University, the National Science Foundation (CBET 1510895, 1547518, and 1802992), and the National Institutes of Health (GM089866).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.