
Abstract
Delivery to the target site and adversities related to off-target exposure have made the road to clinical success and approval of antisense oligonucleotide (AON) therapies challenging. Various classes of AONs have distinct chemical features and pharmacological properties. Understanding the similarities and differences in pharmacokinetics (PKs) among AON classes is important to make future development more efficient and may facilitate regulatory guidance of AON development programs. For the class of 2′-O-methyl phosphorothioate (2OMe PS) RNA AONs, most nonclinical and clinical PK data available today are derived from development of exon skipping therapies for Duchenne muscular dystrophy (DMD). While some publications have featured PK aspects of these AONs, no comprehensive overview is available to date. This article presents a detailed review of absorption, distribution, metabolism, and excretion of 2OMe PS AONs, compiled from publicly available data and previously unpublished internal data on drisapersen and related exon skipping candidates in preclinical species and DMD patients. Considerations regarding drug–drug interactions, toxicokinetics, and pharmacodynamics are also discussed. From the data presented, the picture emerges of consistent PK properties within the 2OMe PS class, predictable behavior across species, and a considerable overlap with other single-stranded PS AONs. A level of detail on muscle as a target tissue is provided, which was not previously available. Furthermore, muscle biopsy samples taken in DMD clinical trials allowed confirmation of the applicability of interspecies scaling approaches commonly applied in the absence of clinical target tissue data.
Introduction
The discovery of oligonucleotides as therapeutic agents unlocked the potential to modulate targets not previously accessible by small molecules or biologics and to address genetic disorders at their root [1]. Oligonucleotide therapies can be divided into various modalities with diverse pharmacological activity. Antisense oligonucleotides (AONs)—single strand oligonucleotides specifically designed to hybridize complementary (pre-)mRNA—may silence mRNA through recruitment of RNase H (gapmers) [2] and by sterical blocking of translation [3], or modulate pre-mRNA splicing, for example, exon skipping and intron inclusion [4]. Other therapeutic applications of oligonucleotides in general include short interfering RNAs (siRNA), silencing mRNA through involvement of the RNA-induced silencing complex (RISC) [5]; aptamers, targeting receptors by their secondary structure [6]; immune stimulating oligonucleotides mimicking foreign RNA [7]; and oligonucleotides mimicking or interfering with micro-RNAs [8].
Recently approved AON therapies have demonstrated the feasibility of their safe therapeutic use [9] and many more are entering late-stage clinical trials [10]. Yet, the road to clinical application in the last decades has not been without hurdles. In particular, the delivery of AON to the site of action and, in connection, adversities related to high (off target) exposure have proved to be challenging [11].
The various classes of AONs have distinct molecular features and chemical modifications [12], and therefore exhibit shared, but also class-specific pharmacological properties, which may also be influenced by sequence and length. Understanding of these similarities and differences is steadily increasing with growing experience in AON drug development documented in public literature. It is foreseen that this information will make future AON development more efficient, for example, facilitating selection of the right modality for the right target, selection of the appropriate preclinical species, sensible study design, and accurate scaling of dosimetry across species [13]. In addition, it may fuel discussions on regulatory guidance for AONs and to what extent requirements should differ from those for small molecules or biologics [14,15].
Most pharmacokinetic (PK) information is available for the class of 2′-O-(2-methoxyethyl) phosphorothioate (2MOE PS) AONs, notably gapmer AONs with a center of DNA nucleotides flanked by 2′ MOE-modified RNA [16,17]. The closely related 2′-O-methyl phosphorothioate (2OMe PS) AONs discussed in this article have a subtle difference in the nature of the 2′ ribose modification and are modified across the full nucleotide length. Most information on 2OMe PS AONs comes from exon skipping therapies developed for Duchenne muscular dystrophy (DMD) [18–27].
DMD is a rare fatal X-linked autosomal recessive disorder characterized by a progressive loss of muscle tissue [28,29]. Drisapersen (PRO051/GSK2402968) was the first exon-skipping therapy for DMD in clinical development [20,21]. It targets exon 51 of the dystrophin gene, with the potential to restore the open reading frame in around 13% of DMD patients (ie, those affected by deletions flanking exon 51). Three follow-up 2OMe AON candidates, targeting exons 44, 45 and 53, were also in clinical development.
Although patients on drisapersen showed improved ambulation in two placebo-controlled phase 2 trials (DEMAND II; DMD114117; NCT01153932 [23] and DEMAND V; DMD114876; NCT01462292 [26]), a pivotal large-scale phase 3 trial with less stringent inclusion criteria (DEMAND III; DMD114044; NCT01254019) did not meet its primary endpoint—a significant improvement in the six-minute walk distance (6MWD) compared to placebo [27]. A post hoc analysis showing significant ambulatory improvement in a subset of younger patients proved not sufficient for regulatory approval in the USA [30]. Subsequently, all four 2OMe PS compounds for DMD were withdrawn from further development.
Despite this setback, the (non)clinical development of these compounds has left a rich dataset that is of interest to the oligonucleotide community. Several articles describe PK aspects of these 2OMe PS AONs, but no comprehensive review was available to date. A large overlap with 2MOE PS AONs is likely, given that the PS backbone is an important driver of PK properties [16]. Although no head-to-head comparisons with the same AON sequence are available, it is of interest to explore similarities and differences between the two related classes.
Furthermore, few other programs have focused on muscle as a therapeutic target tissue. As muscle is neither a tissue of toxicological concern due to lower accumulation compared to liver and kidney, little information on muscle distribution is generally available elsewhere. In the various DMD clinical trials on 2OMe PS AONs, muscle biopsies were routinely taken, providing valuable data on target tissue exposure and effect in relationship to plasma PK. These types of data, which are rarely available in other disease areas, allow to evaluate the interspecies scaling approaches often taken in absence of clinical target tissue samples [31].
This review provides a detailed overview of the PK properties of 2OMe PS oligonucleotides from publicly available sources as well as from unpublished data. Tables and figures presented without literature references contain original, previously unpublished data. Following a brief introduction to oligochemistry, absorption, distribution, metabolism, and excretion, as observed in different preclinical species and in DMD patients, are discussed, as well as the potential for drug–drug interactions (DDIs). Toxicokinetic considerations and pharmacodynamic (PD) aspects, both in general and specific to DMD, are also included. The focus is on drisapersen, the compound with the largest (pre)clinical dataset available to date, where relevant complemented with data on AONs targeting other exons.
Chemistry
Two modifications distinguish 2OMe PS oligonucleotides from endogenous RNA (Supplementary Fig. S1). The hydroxyl group at the 2′ position of the ribose ring is methylated, offering full resistance against degradation by endonucleases (internal cleavage) and an increased melting temperature (Tm) of the duplex with its cognate RNA strand. The nonbridging oxygen in the phosphate group is replaced by a sulfur atom. This PS linkage reduces exonuclease degradation (sequential shortening from the 5′ or 3′ end), but slightly lowers the duplex Tm. The sulfurization turns the phosphodiester moiety into a center of chirality with an Rp and Sp configuration. Using conventional nonstereodefined synthesis, an oligonucleotide of length n has 2n−1 possible stereoisomers. The stereo-configuration of the oligonucleotides impacts functional properties such as hybridization kinetics and metabolic stability.
The closely related class of 2MOE PS AONs differs in the ribose 2′ modification and shows large overlap in chemical and pharmacological properties [16,17]. Depending on the mechanism of action (RNAse H mediated or other), these AONs have 2′ MOE modifications at the flanks only (RNA/DNA chimeric or gapmer) or across the entire length. One fundamental difference is that gapmers are not fully protected by 2′ modifications and hence are subject to internal DNA cleavage by endonucleases.
One key feature driving the PKs of PS AONs is the highly anionic character of the PS linkage. The charge ensures good aqueous solubility and extensive protein binding. In contrast, a chemically distinct class of therapeutically applied AONs, phosphorodiamidate morpholino oligomers (PMOs), has a neutral backbone. The ribose rings are replaced by six-membered morpholine rings, connected by uncharged phosphorodiamidate linkages. Their neutral character profoundly impacts protein binding characteristics, and therefore PKs and safety profile [32].
The optimal length of 2OMe PS AONs, dictated by (intra)cellular distribution kinetics, antisense activity, and tolerability, is typically between 20 and 25 nucleotides. Their size prevents unfacilitated passage of intact cell membranes. Table 1 lists the names, length, and sequence of the 2OMe PS AONs, on which data are presented in this article. Amounts (doses and concentrations) of oligonucleotide in this article refer to their sodium salt forms.
Names, Length (Nucleotides), Sequence, and Molecular Weight (kDa) of 2′-O-Methyl Phosphorothioate Oligonucleotides Referenced in This Article
c*, 5-methylated cytosine.
Absorption
The routes of systemic delivery applied for 2OMe PS AONs have been subcutaneous (SC) and intravenous (IV). Typical plasma profiles of drisapersen upon SC and IV administration in mouse, rat, monkey, and man are shown in Supplementary Fig. S2. The SC absorption is reasonably fast, the time to reach maximum plasma levels depending on species: within 1 h in rodents to around 3 h in monkey and man. Multiple factors may impact the rate of SC absorption, including formulation, concentration/injection volume, and the site of injection. In mice, anesthesia was found to lower the absorption rate [33].
In general, near complete SC absorption is observed in all species. A single exception has been encountered: BMN053 showed an apparent SC bioavailability in DMD patients of around 50% (not shown). The cause of reduced bioavailability of this compound, which showed complete absorption in other species, remains unclear. A metabolic loss of 50% in the course of SC absorption is unlikely. BMN053 potentially shows an unusually high local sequestration, but was not particularly proinflammatory compared to AONs that were completely absorbed.
The four DMD candidates, investigated in clinical trials with doses ranging from 5 to 9 mg/kg, gave rise to maximum plasma concentrations (Cmax), up to 1 μM upon SC administration and 4 μM upon IV infusion (3 h).
Distribution
Plasma protein binding
Like all AONs with a PS backbone, 2OMe PS AONs are promiscuous binders to a range of macromolecules due to their negative charge. They are extensively protein bound in plasma, the main binding plasma proteins being albumin and γ-globulin. In plasma incubation experiments with drisapersen, the fraction bound (as determined by ultrafiltration) was more than 99% across species (Table 2). Some evidence of saturation in mouse and human plasma was observed at the highest concentration tested (95 μM), a concentration much higher than normally achieved in clinical studies. The equilibrium dissociation constant (Kd) of drisapersen with human albumin, as determined by ultrafiltration and isothermal titration calorimetry, was in the micromolar range. While protein binding is extensive, nontarget binding is not strong. AONs are thought to move freely between binding sites and plasma protein binding does not hamper tissue distribution [16].
Fraction of 14C Radiolabeled Drisapersen Bound In Vitro in Male Mouse, Rat, Monkey, and Human Plasma, and Human Albumin, as Determined by Ultrafiltration
The high fraction bound limits glomerular filtration, providing ample opportunity for uptake into tissues. In contrast, oligonucleotides with uncharged linkages such as PMOs show limited plasma protein binding, substantially higher renal clearance, and lower tissue distribution [32].
Plasma profile
Upon IV injection, 2OMe PS AONs like drisapersen display a multiphase plasma concentration-time profile. Plasma concentrations rapidly drop two to three orders of magnitude within hours and elimination then slows gradually before reaching a slow terminal elimination phase. This typical kinetic behavior is illustrated in a radiolabel study (Fig. 1), where groups of CD-1 (wild type) and mdx (dystrophic) mice were sacrificed at various time points up to 100 days after receiving a single IV injection of 100 mg/kg 14C radiolabeled drisapersen. The initial distribution phase reflects rapid binding to cell surface components throughout the body. The AON is then internalized, mainly by endocytosis, redistributed, or excreted. When the bulk of AON has either found its way into the cells or has been cleared, the profile reaches a terminal phase showing equilibrium between tissues and slow washout from tissues into plasma.

Plasma kinetics and tissue distribution after a single IV injection of 100 mg/kg 14C radiolabeled drisapersen in CD-1 (wild type) and mdx (dystrophic) mouse, determined by quantitative whole-body autoradiography (n = 1 per time point). IV, intravenous.
Plasma exposure scales allometrically with body dimensions, with some residual interspecies variation. Drisapersen doses normalized to body weight (mg/kg) gave rise to similar exposure in monkeys and DMD patients, while prediction of human area under the plasma concentration-time curve (AUC) from rodents required an additional scaling factor (Supplementary Fig. S3). Scaling between rodent and human by body surface area (mg/m2) was accurate. A similar relationship between exposure and body dimensions across species was observed for various 2MOE PS gapmer AONs [34].
Tissue distribution
A rapid and wide distribution of AONs throughout the body is seen in all species, but with notable concentration differences among tissues, as illustrated by the whole-body autoradiograms of 14C radiolabeled drisapersen-treated mice in Supplementary Fig. S4. Due to their size and negative charge, 2OMe PS AONs do not passively diffuse across intact cell membranes. The primary route of cellular uptake is by endocytosis. Several cell surface components have been implicated to facilitate uptake, including clathrin, dynamin, stabilin, and caveolin [17,35–38]. Consequently, rather than organ blood flows or membrane transporter expression, tissue distribution is driven by the availability of binding cell surface components and endocytic activity [16,17,39]. The highest concentrations are observed in liver and kidney cortex, followed by kidney medulla, spleen, pancreas, and various other organs.
Muscle tissue typically shows AON concentrations between one and two orders of magnitude lower than liver, making muscle a challenging target to reach. However, a higher uptake is observed in dystrophic muscle (dystrophin-deficient animal models, DMD patients) compared to healthy muscle. For instance, while plasma exposure upon a single drisapersen IV injection of 100 mg/kg was slightly higher in dystrophic mdx mice compared to wild-type CD-1 mice (up to a factor of 2), which was reflected in the concentration difference in many tissues, the difference in skeletal muscle and diaphragm was up to a factor of 8 (Fig. 1). This observation has been confirmed for both 2OMe PS [18] and morpholino AONs [40,41].
Several factors may contribute to the increased uptake by dystrophic muscle. Membrane integrity is compromised in dystrophin-deficient muscle [42]. It has been suggested that the breached integrity of the sarcolemma provides an opportunity for AONs to enter myofibers [18,43,44]. Recent evidence shows that for PMOs, accumulation within muscle tissue almost exclusively occurs in macrophages and satellite cells or myoblasts [45,46]. The merger of these myoblasts delivers PMOs into myofibers in regions of damage and regeneration of dystrophic muscle, mediated directly or indirectly by the presence of macrophages [45–47]. This zonal uptake creates a patchy restoration of dystrophin [48,49].
While for PMOs this seems to be the dominant route of target site delivery—hence the lower muscle accumulation of PMOs compared to 2OMe PS [50]—for 2OMe PS AONs, it may be a route complementary to endosomal uptake and release. 2OMe PS AONs also show much higher accumulation in proliferating and merging myoblasts than in fully differentiated myotubes in vitro. Yet, the dystrophin restoration upon 2OMe PS treatment in vivo has a more uniform appearance (unpublished observations). Despite the low muscle concentrations, the large relative volume of muscular tissue makes it a prominent depot both in wild-type and dystrophic animal models. In mouse, muscle accumulates a higher total amount of AON than liver or kidney.
Brain levels may be detectable upon systemic delivery depending on experimental conditions, but passage of the blood–brain barrier is very limited (Supplementary Fig. S4). For targeting the central nervous system (CNS), AONs must be administered directly into the CNS compartment, for example, by intrathecal or intracerebroventricular infusion. Treatment of neurological conditions such as spinal muscular atrophy and Huntington's disease with AONs by this approach has been proven therapeutically feasible [51–53]. Local administration of oligonucleotides has also been successful in the vitreous cavity of the eye for treatment of retinal conditions [54,55].
Tissue uptake is saturable. With high doses and/or high maximum plasma concentrations, a shift in relative tissue distribution can be observed from highly accumulating organs such as liver and kidney toward tissues that typically show lower uptake. For instance, the effect of route of administration on tissue distribution of PRO052 was investigated in cynomolgus monkey (Fig. 2A, B). The animals received four doses of 6 mg/kg body weight in 7 days by either SC injection or 1 h IV infusion. While SC absorption was complete, evidenced by equal SC and IV plasma AUCs, maximum plasma concentrations—reached at 3 h—were a factor 4 lower than those upon IV infusion. Interestingly, upon IV infusion, concentrations were higher in all muscle-containing tissues, but notably lower in kidney and liver.

Saturation of tissue distribution in cynomolgus monkey depending on route of administration
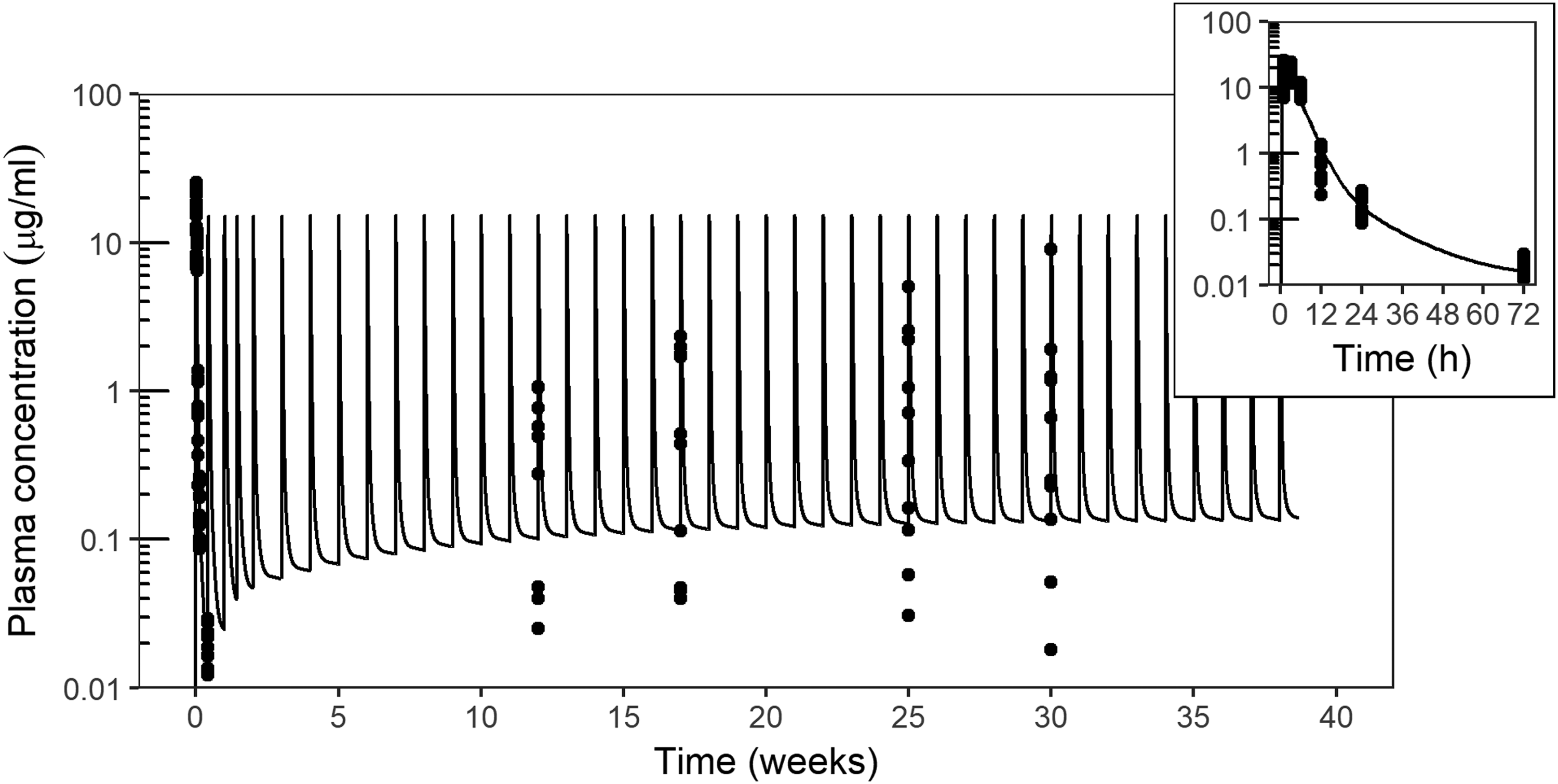
Plasma concentrations of drisapersen (μg/mL, on log10 scale) in cynomolgus monkey receiving SC injections of 6 mg/kg twice weekly for 2 weeks, then weekly for 37 weeks. Symbols mark individual observations at various time points after the first dose and in predose samples at week 12, 17, 25, and 30. The curve represents a PK model simulation. The insert shows a magnification of the profile after the first injection. PK, pharmacokinetic.
Drisapersen metabolism.
A similar pattern was observed in a study investigating the influence of dose and IV infusion duration on tissue distribution of BMN053 in cynomolgus monkey (Fig. 2C, D). Monkeys received IV infusions of 1, 3, or 9 mg/kg twice weekly for 15 days (5 infusions) at an infusion duration of 0.33, 1, 3, or 9 h. One group received 15 daily 0.33 h infusions of 1 mg/kg. Across dose groups with the same infusion duration, a proportional increase in Cmax and a slightly more than proportional increase in AUC were observed. Among the 3 mg/kg dose groups, the plasma AUC drastically decreased with increasing infusion duration. While tissue levels increased linearly between 1 and 3 mg/kg twice weekly at 0.33 h infusion, longer infusions among the 3 mg/kg twice-weekly groups showed a trend toward increasing liver concentrations and decreasing muscle concentrations. Fractionation of the 15 mg/kg cumulative dose into daily doses of 1 mg/kg resulted in a considerable increase in liver uptake and reduction in muscle levels.
These observations indicate that saturation of uptake has both a concentration and a time or frequency component. Possibly, the capacity of cell surface proteins involved in endocytosis can be overwhelmed and needs time to replete. In a drisapersen phase 2 clinical trial, muscle concentrations after 24 weeks of treatment were fourfold higher in patients treated with 6 mg/kg/week compared to 3 mg/kg/week [26].
In contrast, Verhaart et al. [56] found that in mdx mice, fractionation of a weekly dose of 200 mg/kg of m23AON into 100 mg/kg twice-weekly or daily doses of 28.6 mg/kg did lead to an overall higher tissue accumulation, however, without a shift in relative uptake between target and nontarget tissues. At higher plasma exposure level in this study, tissue uptake may have been saturated altogether, possibly increasing renal excretion relative to tissue distribution. Thus, care should be taken to interpret nonlinearities observed in nonclinical species, in relationship to clinically relevant exposure.
The shift in distribution among tissues obtained by adjusting dose, route, rate of infusion, and frequency provides an opportunity to control the safety liability profile (see Toxicokinetic Considerations section) and optimize the therapeutic window of 2OMe PS AONs. Favorable target-to-off-target ratios associated with higher (maximum) plasma exposure may lead to relatively higher efficacy and lower accumulation-related risk (eg, lysosomal accumulation and proinflammatory effects). However, this benefit must be balanced against the increased risk of adversities related to plasma exposure [activated partial thromboplastin time (aPTT), complement activation, and platelet reduction].
Repeated dose, steady-state kinetics
Due to their endosomal storage and slow release, 2OMe PS AONs have a long residence time in tissues. Redistribution among tissues is slow, and it takes time for equilibrium to be reached, as shown in Fig. 1 for mouse and Supplementary Fig. 5 for cynomolgus monkey. Terminal half-lives in tissues are dependent on body dimensions and range from 4–6 weeks in rodents, and 1–2 months in monkey to up to 3–4 months in man.
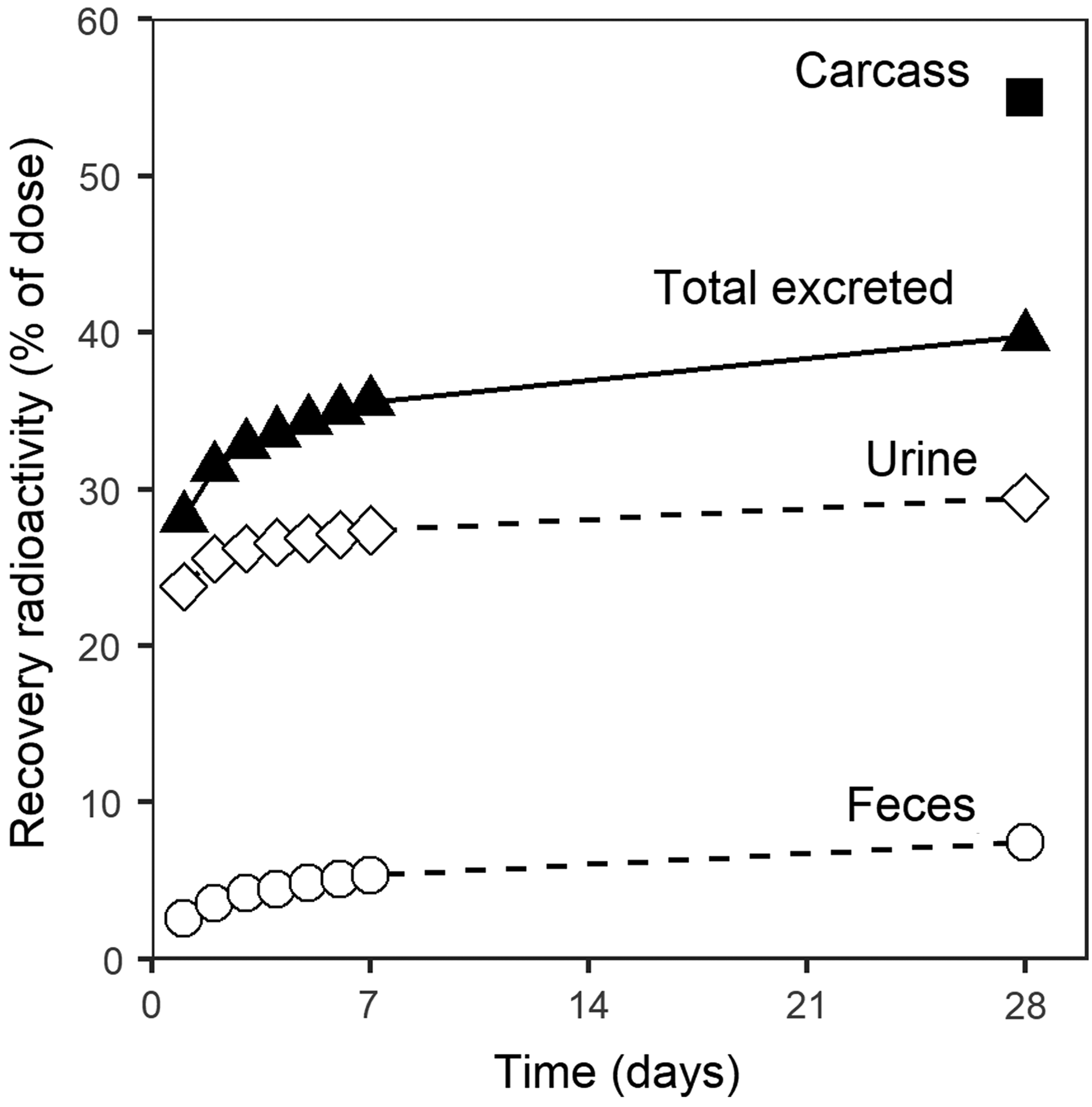
Excretion of radioactivity in CD-1 mouse after a single IV injection of 100 mg/kg 14C labeled drisapersen. Total excreted includes cage wash (amounting to 3% of radioactivity), but not expired air (1.7% in the first 48 h).
With rapid accumulation in tissues and slow release, weekly repeated dosing results in a significant buildup of tissue concentrations. The plasma profile does not reflect tissue concentrations until the terminal elimination phase, as was illustrated in Fig. 1. With weekly dosing regimens, no notable increase in plasma Cmax or AUC is seen over time and evidence of tissue accumulation lies in the end of interval trough plasma concentrations. Figure 3 shows a PK model simulation of drisapersen plasma concentrations over time in a 39-week repeated-dose study in cynomolgus monkey. While plasma Cmax and interval AUC were virtually stable, trough levels rose through the course of treatment toward steady state (reached in ∼7–9 months).
A three-compartment PK model with first-order exchange between central (plasma) and peripheral (tissue) compartments and elimination from the central compartment accurately captured both the plasma concentrations in time after the first administration and the increase in plasma trough levels after repeated dosing. Note that the increased variance in trough concentrations and the slight underestimation of median trough levels by the model at later time points indicate the potential rise of binding antidrug antibodies (see Toxicokinetic Considerations section). The half-life of trough level accumulation toward steady state corresponds to observations of tissue half-life. This implies that the exchange between tissues and plasma obeys first-order PKs.
Metabolism
Where the introduction of PS provides some protection, 2′-O methylation makes AONs fully resistant to endonuclease degradation. So, in contrast to first-generation AONs (full PS) and gapmers (PS backbone with 2′ modifications at the flanks only) [16,17], no internally cleaved products of 2OMe PS arise in vivo [13]. Metabolism takes place by exonucleases, sequentially cleaving the terminal PS bond almost exclusively from the 3′ end. Depending on experimental conditions, 5′ shortened metabolites may be detectable in some tissues, but they generally comprise a negligible fraction of total drug.
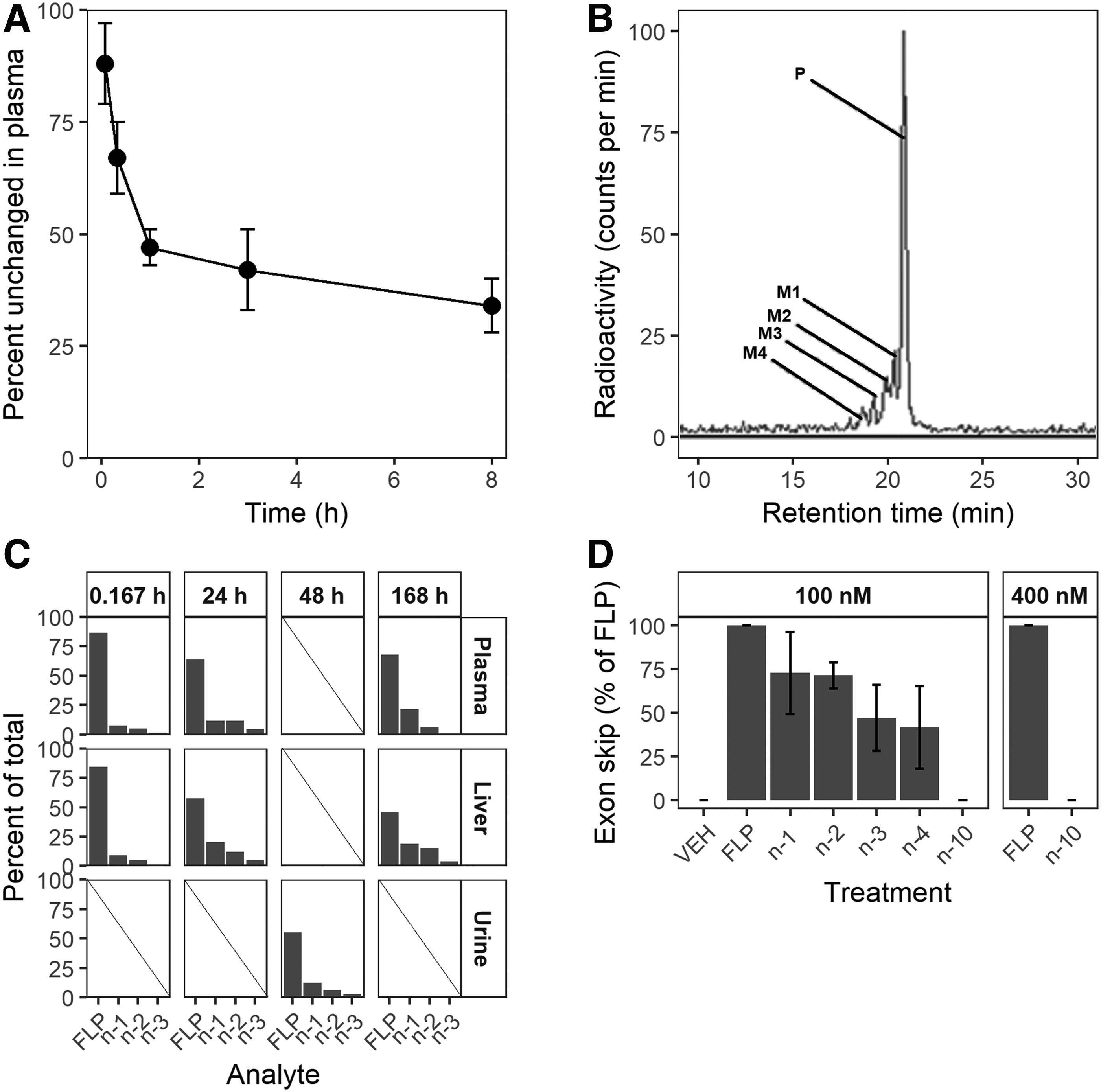
Exonucleases are located in sera and in the intracellular space of diverse tissues [57]. Consequently, 3′ degradation starts directly upon parenteral administration and continues after internalization into tissue. The initial rate of metabolism can be quite rapid. Figure 4A depicts the percent of unchanged drisapersen in plasma over time in mice. A substantial fraction of the full-length product (FLP) is degraded into shorter metabolites within 10 min after IV injection, at an estimated initial half-life of 15 min. The rate of metabolism then declines and levels off at just below 50%.
A radiolabel study in CD-1 mice showed a similar picture (Fig. 4). The radiochromatogram of a plasma sample taken at 24 h post-IV injection of 100 mg/kg 14C radiolabeled drisapersen (Fig. 4B) shows that the largest fraction is present as unchanged drug, with 3′ n-1 to n-4 metabolites detected at decreasing fractions. The fractions of metabolites in plasma increased from 10 min to 24 h, but remained stable for 6 days after that (Fig. 4C). Similar metabolite profiles were observed in the liver and in urine.
This triangular shape profile of FLP and 3′ shortened metabolites, typically observed with fully 2′ modified PS AONs, is a consequence of the chirality of the PS bond. Exonucleases are stereoselective: Rp linkages are cleaved off rapidly, while Sp linkages are much more resistant to exonuclease activity [58–60]. The extent of metabolism thus depends on the fraction of Rp and Sp linkages at the first positions from the 3′ end. The stereorandom synthesis of drisapersen and other DMD exon AONs typically yielded around 60% Rp, with some dependence on the specific nucleotides linked. Indeed, fractions metabolized approaching 60% have been observed in animal studies and clinical trials (unpublished results). The rate of metabolism may differ between compounds, depending on the 3′ terminal sequence and nucleobase modifications (eg, methylation of cytosines). The metabolic profiles observed in mice, monkeys, and human subjects are similar. Thus, in general, preclinical safety studies provide good metabolite exposure coverage.
Metabolites retain some affinity for their target, and efficacy. In incubation experiments using a DMD patient cell culture, the exon skip efficiency of 3′ metabolites of drisapersen at 100 nM gradually decreased to around 40% of FLP for the n-4 (16mer) (Fig. 4D). The 10mer 3′ n-10 showed no exon skip activity even at 400 nM. Given the rapid convergence to a fixed profile of metabolites and their incremental decrease in activity, metabolism only has a mild impact on efficacy and a sufficient understanding of the PK/PD relationship is usually obtained by observing just the parent PK. This may not apply to conjugated AONs or AONs with specific (3′) terminal modifications: cleavage or conversion of the modified termini may then result in a significant change in PK and/or PD behavior.
Excretion
Extensive protein binding of 2OMe PS AONs limits renal clearance. Still, renal excretion is the dominant route of elimination. Figure 5 summarizes the result of a mass balance study of 14C labeled drisapersen in CD-1 mice. After 28 days, the largest fraction of administered AON was still present in the carcass. Most of the excreted AON was found in urine (29% of the dose), and a smaller fraction in feces (7%). A substantial fraction of renally cleared radioactivity was excreted in the first 24 h, when the AON is mainly bound to cell surface components, but not yet fully taken up into cells. The rate of excretion then declined, reflecting the slow elimination of AON from tissues once endocytosed. Fecal excretion was more gradual, which logically follows from the time required for AONs to cross the enteric barriers. No enrichment of metabolites was observed in excreta relative to plasma (Fig. 4C), indicating no gross alteration in elimination kinetics of 3′ shortened metabolites.
Overall, monitoring excretion of several AONs in preclinical mass balance studies has not revealed unexpected behavior. The pattern of excretion closely mirrors observations of plasma PK and tissue distribution.
Drug–Drug Interactions
Oligonucleotides in general are not substrates for cytochrome P450 (CYP) or phase 2 metabolic enzymes [uridine 5′-diphospho-glucuronosyltransferase (UGT) and sulfotransferases], nor for the major membrane transporter proteins. Thus, AON distribution and metabolism are not likely altered in the presence of small molecule substrates or inducers of these enzymes and transporters.
AONs could however potentially interfere as perpetrators with CYPs or phase 2 enzymes by aspecific binding to intracellular proteins. To achieve this, AONs would have to reach sufficient free concentrations in cytoplasm. Their uptake through endocytosis, slow release, and promiscuous binding to macromolecules make that unlikely. Drisapersen did not show any potential for inhibiting the enzyme activity of the most important cytochrome P450 enzymes [61,62] toward their model substrates in vitro. Figure 6A shows the results of primary human hepatocyte incubations with substrates of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5 and drisapersen or respective positive controls for direct or metabolism-dependent inhibition. While the test system showed the appropriate response to the various positive controls, neither concomitant incubation nor 30-min preincubation with drisapersen at concentrations up to 135 μM had any relevant effect on model substrate turnover.
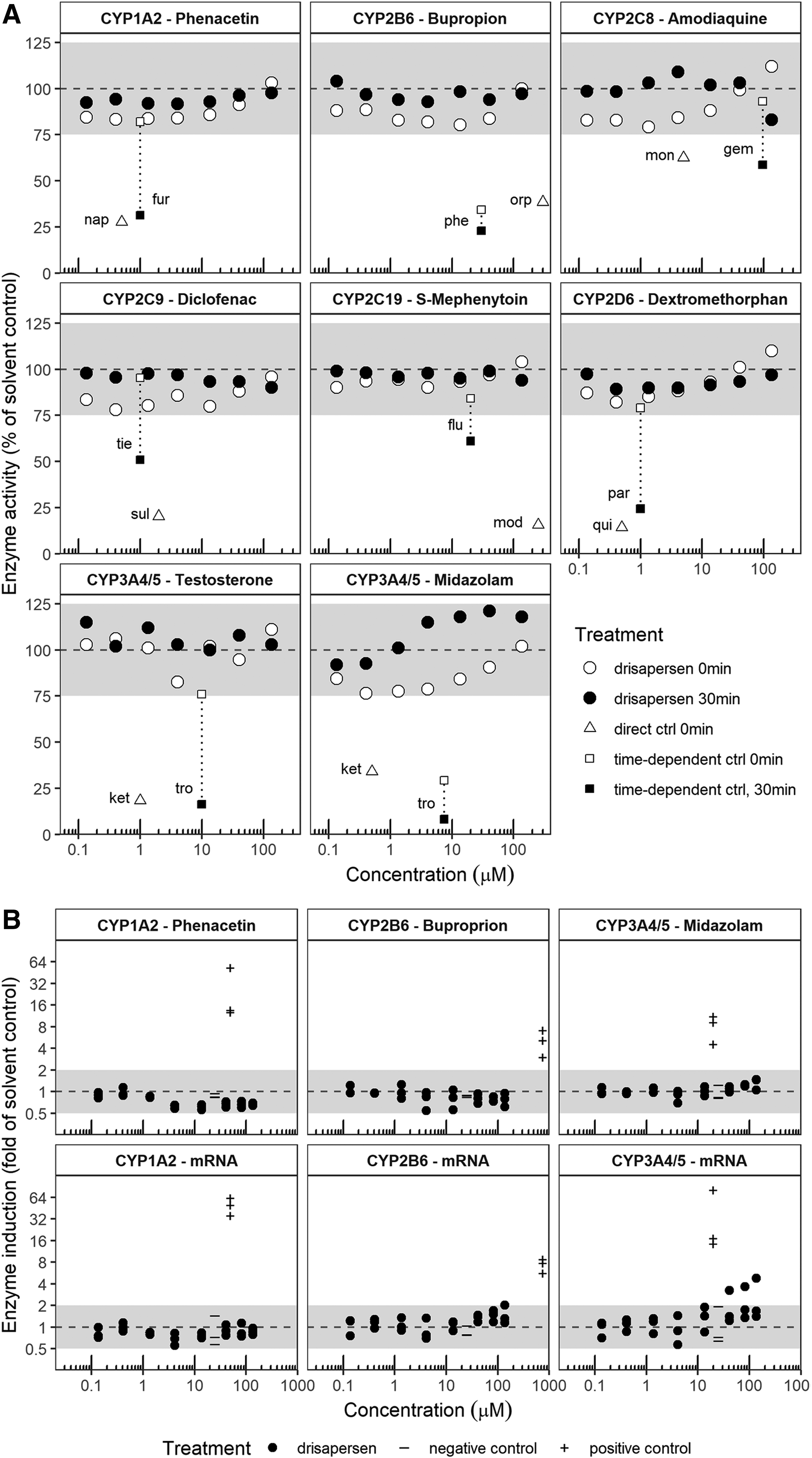
In vitro cytochrome P450 drug–drug interaction studies with drisapersen.
Figure 6B demonstrates the lack of relevant enzyme induction by drisapersen in primary human hepatocytes in vitro. Treatment of cultured human hepatocytes with up to 135 μM drisapersen had little or no effect on CYP1A2, CYP2B6, or CYP3A4/5 activity (within twofold of solvent controls and below 20% of the positive control). No effect was also seen at the mRNA level, with two exceptions. Treatment with 135 μM drisapersen caused a 1.03-fold increase in CYP2B6 mRNA, while treatment with 40.6 to 135 μM drisapersen caused concentration-dependent increases from 2.27- to 3.83-fold in CYP3A4 mRNA, both in one out of three donors. However, these increases were <15% as effective as the appropriate positive controls and had no apparent effect on substrate turnover, and the estimated concentration of half-maximum inhibition (IC50) for CYP3A4/5 induction was far above clinically relevant concentrations (R3 above 0.9 [62]).
In accordance with our findings, three other PS backbone AONs (two gapmers and one uniform 2MOE) and one GalNAc conjugated mixed PO/PS AON have been shown not to interfere with CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5 metabolism in cryopreserved human hepatocytes in vitro at concentrations up to 100 μM [63]. Kazmi et al. found that two PS AONs without 2′ ribose modification, but not phosphodiester AONs of the same sequence, inhibited various CYPs and UGTs in human liver microsomes with EC50s ranging between 1 and 100 μM [64]. In cryopreserved human hepatocytes, however, similar concentrations did not inhibit the same enzymes. These results highlight the importance of the right cellular context of in vitro DDI studies. Clinical DDI studies of various PS AONs with likely concomitant medications to date have not revealed any relevant interactions [65–69].
Oligonucleotides have not been identified as substrates for the main drug transporter proteins [63]. With low affinity binding to proteins in general, they are unlikely to show relevant interference with drug transporter activity. Several PS backbone AONs were found to not interfere with a panel of six influx and three efflux transporters at a concentration of 10 μM [63,70]. The two PS AONs without 2′ modification investigated by Kazmi et al. were able to inhibit the influx transporters OATP1B3, OAT1, OAT3, and OCT2 in vitro with IC50s ranging from 12 to 92 μM, of which one also inhibited OATP1B3 with an IC50 of 90 μM [64]. OCT1 and the efflux transporters P-gp and BCRP were not affected at concentrations up to 100 μM. When interpreting the in vitro inhibition of apical drug transporters in particular, the fraction bound should be taken into account [62,71,72]. PS AONs are extensively bound in plasma, and only the fraction of drug that is not bound to other proteins may interfere with transport. If at all present, protein concentrations in in vitro incubation media are typically orders of magnitude lower than in plasma. Corrected for protein binding, clinical plasma AON concentrations are unlikely to come near those that may interfere with transporter activity.
The potential of PS AONs to inhibit CYPs and transporters is based on aspecific protein binding, driven by backbone chemistry. The observed lack of inhibition at clinically relevant concentrations is therefore considered representative for the entire class.
Relevant DDI due to plasma protein binding displacement is unlikely. The negatively charged PS AONs bind preferentially to Sudlow site I of human serum albumin [73], implicating warfarin-like, acidic drugs as potential victims of displacement. However, maximum human exposure to AONs is typically in the low micromolar range, much lower than the plasma concentration of albumin (∼600 μM). Furthermore, while AONs are extensively bound in plasma, the association with plasma proteins is relatively weak, with dissociation constants in the micromolar range. 2OMe PS AONs are equally unlikely to be the victim of protein binding displacement. Potential displacement by a strong binder to albumin would leave a host of other macromolecules to bind with sufficient affinity. Various PS backbone AONs have indeed been demonstrated to not show displacement interactions in human plasma in vitro at clinically relevant concentrations [74]. No clinical interactions were observed between the PS AON mipomersen and warfarin [69].
Toxicokinetic Considerations
A well-defined array of safety concerns is commonly associated with 2OMe PS AONs in preclinical and/or clinical studies, inherent to their chemistry and PK properties, including their DNA-/RNA-like nature, promiscuous plasma protein binding, and endosomal sequestration in tissues. Some effects associated with the PS backbone can be considered class effects [13,75,76]. For these class effects, a wide overlap is commonly observed between 2OMe and 2MOE PS, given their common backbone and subtle difference in 2′ modifications. Other effects have a stronger dependency on sequence. Table 3 presents a summary of the toxicokinetics associated with common safety concerns discussed in more detail below.
Toxicokinetic Considerations Regarding Potential Safety Concerns of 2′-O-Methyl Phosphorothioate Antisense Oligonucleotides
AON, antisense oligonucleotide; aPTT, activated partial thromboplastin time; IV, intravenous; SC, subcutaneous.
A concern specific to oligonucleotide therapies is the potential to alter protein expression by off-target hybridization. The risk is limited or prevented by careful design, in silico screening for sequence complementarity against the human genome [77]. For 2OMe PS AONs with a typical length of 20–25 nucleotides that act by modulation of splicing or steric hindrance, the likelihood of potential off-target complementary mRNA stretches is low. For drisapersen (a 20mer), a BLAST against the human genome did not identify any alternative mRNA targets, even when multiple mismatches were allowed. The increased likelihood of off-target complementarity with sequentially shortened metabolites is counterbalanced by progressively lower concentrations and reduced binding affinity, so that off-target hybridization-dependent toxicity at clinically relevant concentrations remains highly unlikely. Hybridization-dependent effects have been described for RNase H1 activating gapmer AONs, notably the short versions with hybridization enhancing modifications [78–80] and for siRNA [81,82]. These modalities contract enzyme complexes to cleave mRNA, which seem to have some tolerance for mismatches. For these AON classes, additional in vitro and in vivo screening strategies have been proposed to mitigate the off-target hybridization risk [83–85].
A further consequence of the DNA-/RNA-like nature of PS oligonucleotides is their tendency to be recognized as foreign by the innate immune system. Certain sequence motifs in particular (unmethylated CpG motifs) were found to be potent immunostimulants through recognition by Toll-like receptors, notably TLR9 [86]. In current AON design, such immunostimulatory motifs are avoided if possible. Methylation of the ribose 2′O and of cytosines have further reduced, but not fully eliminated, the proinflammatory potential of 2OMe PS AONs. Innate immune stimulation can be considered a class effect to some extent, but with strong dependence on sequence. The inflammatory response is concentration dependent; thus, obvious targets include the injection site, liver, and kidney. Inflammatory lesions may progress beyond steady state [87]. The long tissue half-life implies a dependency on duration of treatment, and slow resolution upon cessation of treatment. The manifestation of AON-induced inflammation differs between species, with rodents being more sensitive than primates [88,89].
Plasma protein binding may affect blood clotting and complement cascades. Reversible binding to components of the tenase complex leads to prolongation of the aPTT, which can be observed in all species [90,91]. Binding to complement factor H causes activation of the complement alternative pathway [92,93]. Monkeys have shown to be considerably more sensitive to complement activation than humans [88,89,94–96]. Both effects are strongly correlated with plasma exposure, and thus show an acute onset upon administration and rapid restoration to baseline as the AON disappears from plasma. Inflammatory lesions secondary to prolonged complement activation, which may include glomerulonephritis or vasculitis, take longer to resolve [88,89,95]. The clinical risk is managed by keeping the Cmax below the respective monkey thresholds, either by SC administration or slow IV infusion, and monitoring aPTT and complement split products (C3a, C5a, and Bb) directly after administration.
Lysosomal accumulation in tissues may lead to histological changes, apparent as basophilic granules. These are most prominent in kidney cortex (proximal tubules) and liver [89]. Histological changes occur in a dose-dependent manner, with a gradual increase with duration of treatment until steady state is reached. Pathological manifestations, including elevated liver damage markers in serum and proteinuria, have a high threshold and usually remain subclinical [95]. Full recovery is seen upon cessation of treatment, correlated with tissue half-life.
Some PS AONs induce a mild and gradual decrease in mean platelet count upon repeated dosing, stabilizing into a steady state within the normal range (above 150 × 109platelets/L). This mild platelet reduction is observed across species and affects all animals in a dose-dependent manner. Platelet reduction is not considered a class effect, as it has been observed in preclinical and clinical studies only in subset of PS AONs studied [96,97]. Investigations closing in on the mechanism of action are ongoing. Above a certain threshold of 5 mg/kg/week in nonhuman primates and humans, sporadic cases have been reported of a more precipitous drop in platelet count to levels below 50 × 109platelets/L [96–98]. The distinct pattern, low incidence, and lack of reproducibility suggest a separate mechanism of action, potentially involving interaction with a predisposed background [89,96]. In both the mild gradual and the more severe progressive phenotype, cessation of treatment restores platelet counts to baseline levels.
The PS backbone of AONs has the potential to induce an antidrug antibody (ADA) response. The incidence of immunogenicity depends on dose and duration of treatment. Approximately 25% of patients in drisapersen clinical trials up to 48 weeks of treatment were found ADA positive. Titers may rise with continued exposure before typically stabilizing. Characterized by binding antibodies, the immunogenic response is unrelated to other immune stimulatory properties of AONs (innate immune response and complement activation). In drisapersen clinical trials (nearly 300 patient-years of treatment analyzed), no correlation was found between ADA presence or titer and adverse events or safety biomarkers, including inflammatory cytokines, complement C3, thrombocyte count, and injection site reactions.
The presence of ADA not only in patients but also in nonclinical studies in monkey is indicated by a more than linear rise of plasma trough levels: increased plasma binding to ADA without neutralization decreases renal clearance and increases plasma residence time. An example of such elevated plasma trough levels in a suspected ADA-positive monkey is shown in Supplementary Fig. S6. The prolonged plasma residence time widens the window for uptake into tissue. Although not evident when comparing collective average muscle biopsy concentrations between ADA-positive and ADA-negative DMD patients in drisapersen trials, population PK analysis revealed a significant increase in target tissue concentrations with increasing ADA titer.
PK/PD Considerations
While AONs are taken up into cells predominantly by endocytosis, their molecular target is located either in the nucleus (pre-mRNA modulation and exon skip) or cytoplasm (RNA degradation and sterical blocking of RNA translation). In both cases, the AON first needs to be released from the endosome to allow it to reach its molecular target. Once in the cytoplasm, AONs efficiently distribute to other cell compartments, including the nucleus [99,100]. In vitro co-incubations with transfection agents to avoid endosomal entrapment (eg, polyethylenimine) or with stimulants of endosomal release result in a vast increase in nuclear AON concentration and efficacy compared to unaided (gymnotic) uptake (unpublished observations).
The PK/PD relationship of AONs in general is characterized by a fairly rapid target engagement and onset of effect at (pre-)mRNA level. This also applies to exon skipping 2OMe PS AONs, as demonstrated in dystrophic mice (mdx) in Fig. 7. The onset and duration of mRNA modulation closely followed the observed concentration of AON in (total) tissue from the exposure phase through washout. This suggests that dystrophin mRNA turnover has a half-life shorter than or similar to tissue AON, and that a fixed proportion of total AON is present at the site of action throughout exposure. Thus, AON is continuously released from the endomembrane compartment into cytoplasm and/or nuclei. Translation into dystrophin protein takes time, as indicated by the delay in protein level effect. Dystrophin half-life may vary with circumstances such as the specific exon deletion involved and muscle type, but the duration of effect is consistently similar to or longer compared with mRNA. This mirrors clinical observations, in which the observed improvements in the clinical endpoint (the 6MWD) after 24 weeks of treatment were largely maintained during a 24 week off-treatment period [26].
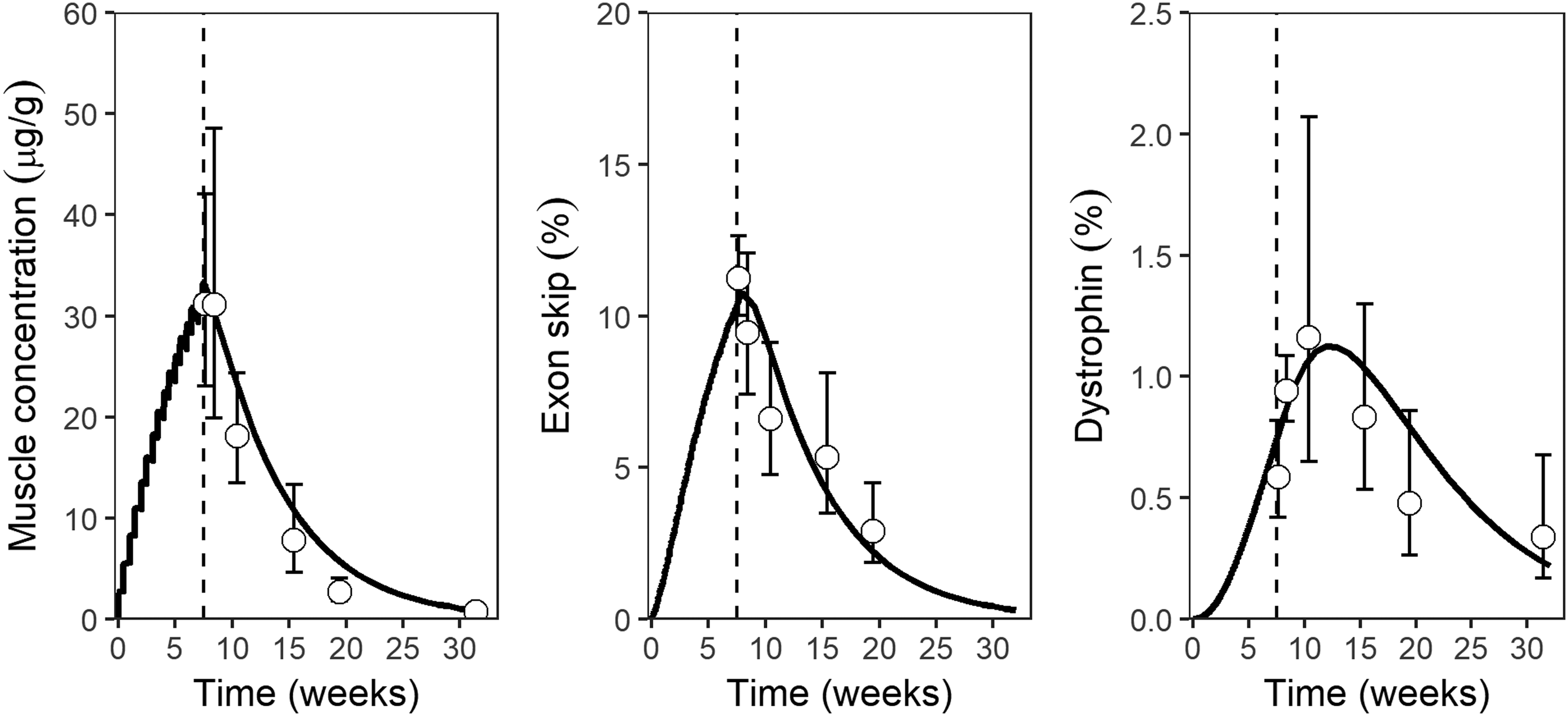
PK/PD relationship of m23AON, a mouse dystrophin exon 23 skipping 2OMe PS AON, in the mdx mouse model. The panels show the quadriceps muscle concentration (μg/g), exon skip (% of transcripts skipped, determined by RT-PCR), and dystrophin protein (% of normal, determined by western blot) against time (weeks) after start of treatment of 100 mg/kg m23AON twice weekly for 8 weeks by SC injection. Symbols and error bars indicate observed geometric means and standard deviations (n = 6), curves represent simulations by a PBPK/PD model. The dashed line marks the end of treatment. The data were published earlier in modified form in Verhaart et al. [19]. 2OMe PS, 2′-O-methyl phosphorothioate; PBPK, physiologically based pharmacokinetic; PD, pharmacodynamic; RT-PCR, reverse transcriptase polymerase chain reaction.
DMD is an X-linked disorder almost exclusively affecting boys. Given the disease progression, exon skipping therapy should be started as soon as possible after diagnosis. This makes age and gender two important factors to consider in DMD drug development. 2OMe PS AONs are not substrates for CYPs, phase 2 conjugation enzymes, or major drug transporters. Therefore, profound changes in PK with age related to the ontogeny of such proteins, sometimes observed with small molecule therapeutics, are not expected for AONs. Age may impact their PK in a more gradual way, consistent with changes in physiology. For instance, differences in plasma exposure to drisapersen between juvenile and adult CD-1 mice were in line with the allometric relationship with body weight observed between species. Furthermore, disease progression with age affects muscle uptake, as discussed further below. 2OMe PS AONs in development for DMD have not shown any relevant gender differences in PK properties in preclinical studies, other than those attributed to differences in body dimensions. Mixed-gender preclinical studies, which may be more convenient than male only, have generally yielded similar results.
With target tissue samples from patients not commonly available and tissue exposure so distinct from the plasma profile, understanding the clinical exposure-response relationship of AONs acting in tissues can be a challenge. If the target is a circulating protein or has a circulating biomarker, insight in target tissue exposure can be gained through dose-response-time analysis [101]. A more common strategy is to estimate tissue concentrations from trough levels using the tissue-to-plasma trough ratio obtained in preclinical studies. This ratio has been found to be fairly constant for several 2MOE PS AONs between mouse and monkey [17,31].
In the DMD clinical programs, muscle biopsies were sampled, which provide more information on target site exposure and effect than available in many other disease areas and allow to verify the scaling approach against clinical data. Figure 8 plots the muscle-to-plasma trough concentration ratios observed in wild-type (CD-1) and dystrophic (mdx) mice, monkey, and DMD patients. Indeed, the muscle-to-plasma trough ratio is constant between healthy mouse and monkey. In dystrophic mice (mdx model) and DMD patients, the ratios are nearly a factor of three higher due to increased muscle uptake. Accounting for the higher distribution to dystrophic muscle, muscle concentrations can thus be accurately estimated from plasma trough concentrations by preclinically determined ratios.

Drisapersen muscle-to-plasma trough concentration ratios across species.
Even with the availability of DMD muscle biopsies, it remains challenging to accurately estimate the long tissue half-lives in patients. Washout phases in clinical trials do not necessarily extend to the duration required for reliable estimation of terminal half-life: four to five AON half-lives may equal over 1 year. Plasma concentrations as a surrogate may not remain above limits of quantification during that time. Muscle half-life estimated for drisapersen based on tissue concentrations observed in 24-week washout periods was considerably lower than that estimated by population PK modeling. The latter made use of additional information, including trough plasma concentrations over durations of exposure longer than the washout phase. Even if plasma elimination may appear approximately linear, some redistribution among tissues may still be occurring. Thus, AON terminal elimination half-lives estimated from short washout phases must be interpreted with caution.
DMD is characterized by a progressive loss of muscular tissue through repeated cycles of muscle damage, necrosis, and regeneration [28]. The decreasing relative volume of muscular tissue and loss of regenerative capacity may contribute to the decrease in muscle AON concentrations with age observed in various drisapersen clinical trials shown in Fig. 9A. The reduced muscle uptake with age and/or body weight is accompanied by an increase in plasma AUC (not shown).

(
The muscle AON concentration in turn correlates with clinical benefit. Drisapersen treatment in clinical trials gave rise to low levels of exon skip and dystrophin. In the pivotal clinical trial [27], the primary endpoint—the 6MWD—did not reach significance when comparing treatment against placebo. Yet, a post hoc meta-analysis across studies did reveal a clear tissue concentration–response in 6MWD (Fig. 9B). These findings underline that the earlier in disease progression the treatment starts, the more beneficial exon skipping therapy is. This is likely explained by a higher AON accumulation in muscle and a higher susceptibility to dystrophin rescue.
Conclusions
Overall, 2OMe PS AONs of different length and sequence demonstrate comparable PK properties. Some PK properties observed with drisapersen, such as the profile of metabolism and excretion and the lack of in vitro DDI, can be considered representative for the entire class and should allow to waive particular studies into those properties for future 2OMe PS AONs.
From the collective data presented, a high degree of similarity in PK emerges between the fully modified 2OMe PS and other PS backbone AONs, including 2MOE gapmers.
The PK behavior is consistent and predictable across species, with few exceptions. Preclinical studies in a DMD mouse model and cynomolgus monkey gave a complete picture of the PK of drisapersen that accurately predicted its behavior in DMD patients.
DMD clinical studies provided muscle biopsy samples, allowing to confirm the applicability of the interspecies scaling strategy commonly applied in the absence of clinical target tissue data. Ratios between plasma trough levels and muscle tissue were constant across species after accounting for higher uptake in dystrophic versus healthy muscle.
With muscle accumulation one or two orders of magnitude lower than that in liver and kidney cortex, muscle is a challenging tissue to target with AON therapy. Dose limiting toxicity prevented higher drisapersen muscle levels to be reached in patients. For future exon skipping AONs for DMD with more specific muscle targeting, higher efficacy, and/or an improved safety profile, there may be room for optimizing the dosing regimen. Preclinical studies appeared to favor IV over SC administration, and faster infusion rates, but the benefits should be balanced against increased safety liabilities related to maximum plasma exposure.
Exon skipping therapy for DMD has been most beneficial to younger patients, likely explained by a higher availability of viable muscle to rescue and a higher uptake of AON in regenerating muscle at early disease stages.
Oligonucleotide therapies have gained momentum with recent clinical success and approvals [9]. Although drisapersen did not gain regulatory approval, it has provided a rich preclinical and clinical dataset. Insight into the PK properties of 2OMe PS AONs in this review may be of benefit to future developments in this class of AONs, serve as input for discussions around regulatory requirements for oligonucleotides [14,15], or serve as a benchmark for chemical modifications to improve PKs, efficacy, and therapeutic window, in the DMD disease area and beyond.
Footnotes
Acknowledgments
This article was based on collective efforts of (former) colleagues at BioMarin Pharmaceutical, Inc., Prosensa Therapeutics BV, Leiden University Medical Center, GlaxoSmithKline, and numerous collaborators. Fruitful discussions in the Oligonucleotide Safety Working Group subcommittee on DMPK helped shape this review.
Author Disclosure Statement
N.D. and J.v.D. are current and S.B., J.S., S.d.K., and C.d.B. are former employees of BioMarin Nederland B.V., which developed drisapersen and related oligonucleotides for DMD.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.