
Abstract
Inotersen, a 2′-O-methoxyethyl (2′-MOE) phosphorothioate antisense oligonucleotide, reduced disease progression and improved quality of life in patients with hereditary transthyretin amyloidosis with polyneuropathy (hATTR-PN) in the NEURO-TTR and NEURO-TTR open-label extension (OLE) trials. However, 300 mg/week inotersen treatment was associated with platelet count reductions in several patients. Mean platelet counts in patients in the NEURO-TTR-inotersen group remained ≥140 × 109/L in 50% and ≥100 × 109/L in 80% of the subjects. However, grade 4 thrombocytopenia (<25 × 109/L) occurred in three subjects in NEURO-TTR trial, and one of these suffered a fatal intracranial hemorrhage. The two others were treated successfully with corticosteroids and discontinuation of inotersen. Investigations in a subset of subjects in NEURO-TTR (n = 17 placebo; n = 31 inotersen) and OLE (n = 33) trials ruled out direct myelotoxicity, consumptive coagulopathy, and heparin-induced thrombocytopenia. Antiplatelet immunoglobulin G (IgG) antibodies were detected at baseline in 5 of 31 (16%) inotersen-treated subjects in NEURO-TTR, 4 of whom eventually developed grade 1 or 2 thrombocytopenia while on the drug. In addition, 24 subjects in the same group developed treatment-emergent antiplatelet IgG antibodies, of which 2 developed grade 2, and 3 developed grade 4 thrombocytopenia. Antiplatelet IgG antibodies in two of the three grade 4 thrombocytopenia subjects targeted GPIIb/IIIa. Plasma cytokines previously implicated in immune dysregulation, such as interleukin (IL)-23 and a proliferation-inducing ligand (APRIL) were often above the normal range at baseline. Collectively, these findings suggest an underlying immunologic dysregulation predisposing some individuals to immune-mediated thrombocytopenia during inotersen treatment.
Introduction
Hereditary amyloidosis is a disease that results from misfolding and deposition in tissues of various mutated proteins, such as transthyretin (TTR), as amyloid fibrils that interfere with the normal functioning of numerous organs [1]. TTR is an abundant soluble serum protein produced predominantly in the liver that transports both vitamin A (via retinol-binding protein) and thyroxine. Hereditary TTR amyloidosis (hATTR), the most common hereditary amyloidosis, is a rare, autosomal-dominant, progressive, and fatal disease caused by point mutations in the TTR gene [2,3]. These mutations destabilize the tetrameric TTR protein complex, which tends to dissociate into monomers that subsequently denature and misassemble into a spectrum of aggregate structures including amyloid fibrils [1,4]. Amyloid fibrils are then deposited as insoluble, extracellular amyloid deposits in multiple tissues and organs, including the heart, peripheral nerves, kidney, gut, meninges, and eyes leading to progressive peripheral sensorimotor and autonomic polyneuropathy, cardiomyopathy, nephropathy, blindness, brain ischemia, and gastrointestinal dysfunction [5].
Inotersen, a 2′-O-methoxyethyl (2′-MOE) phosphorothioate antisense oligonucleotide (ASO), is an approved treatment for polyneuropathy of hATTR (hATTR-PN) in the United States, Europe, and Canada [6]. Inotersen hybridizes with liver TTR mRNA transcript and reduces mRNA levels through an RNaseH1 mechanism of action, leading to reductions in both mutant and wild-type TTR protein [7]. In a recently completed multinational, randomized, placebo-controlled phase 3 study (NEURO-TTR; NCT01737398) in 172 subjects with hATTR-PN, inotersen significantly reduced the progression of polyneuropathy and improved quality of life compared with placebo [8].
Thrombocytopenia was the principle safety concern during NEURO-TTR and NEURO-TTR open-label extension (OLE) studies. Some subjects had significant platelet count decreases, including three cases of confirmed grade 4 thrombocytopenia (<25 × 109/L), one of which presented with a fatal intracranial hemorrhage. All three cases occurred while platelet monitoring was carried out every 6 weeks and before the implementation of weekly testing of platelet counts. Reduced platelet counts were rapidly reversible in two subjects with grade 4 thrombocytopenia after drug discontinuation and administration of corticosteroids. Therefore, experimental investigations focused largely on understanding potential immune-based mechanisms that could have played a role in contributing to inotersen-induced grade 4 thrombocytopenia. Specifically, immune-mediated mechanisms that could involve either antiplatelet factor 4 (anti-PF4) immunoglobulin G (IgG) antibodies because of the polyanionic nature of ASOs [9] potentially resulting in a heparin-induced thrombocytopenia (HIT)-like syndrome, or the presence of drug-induced antiplatelet antibodies [10] were investigated. We also performed laboratory testing for potential biomarkers in plasma from all subjects in NEURO-TTR at baseline to try to identify biomarkers for hATTR-PN subjects who may be at risk for grade 4 thrombocytopenia during treatment with inotersen.
Materials and Methods
Subjects
NEURO-TTR was a phase 3, multicenter, double-blind, randomized, placebo-controlled study. Eligibility for participation in the OLE study (NCT 02175004) required the satisfactory completion of treatment in NEURO-TTR. Subjects receiving inotersen in NEURO-TTR continued to receive inotersen in the OLE study and are hereafter referred to as the inotersen-inotersen group. Subjects receiving placebo in NEURO-TTR switched to inotersen in the OLE study and are hereafter referred to as the placebo-inotersen group.
Laboratory investigations
Detection of anti-PF4 antibodies and drug-dependent/independent antiplatelet antibodies in serum, analysis of biomarkers in plasma, and collection of routine hematology and clinical chemistry parameters were performed. In addition, complement activation [measurement of complement (C3, C4) and split products (Bb and C5a)] assays, and in vitro platelet activation (P-selectin; CD62P upregulation on platelet surface using flow cytometry) tests were also performed, results of which are discussed in Supplementary Data, Supplementary Table S1 and Supplementary Figure S1.
Exploration of potential immunologic causes for thrombocytopenia in NEURO-TTR and OLE studies
To investigate the potential for an inotersen-induced HIT and/or antiplatelet antibody response, serum from 49 (17 placebo and 32 inotersen subjects) of 172 subjects treated in NEURO-TTR study and 33 (10 in placebo-inotersen arm, 23 in inotersen-inotersen arm) of 114 subjects enrolled in OLE study were tested (Supplementary Tables S2–S4) for presence of heparin-anti-PF4 IgG/IgM/IgA and antiplatelet IgG/IgM antibodies (drug dependent/independent) at Blood Center of Wisconsin (Milwaukee, WI). Subjects whose samples were antiplatelet IgG positive were tested further to identify putative glycoprotein binding sites (eg, GPIIb/IIIa) for these antibodies on the platelet. Twenty-nine subjects tested in both NEURO-TTR and OLE studies were selected in a blinded manner during the pivotal study. Subjects tested included 20/26 inotersen-treated subjects in NEURO-TTR who had a confirmed nadir platelet count <100 × 109/L, 10 of 11 subjects in NEURO-TTR who were dose-paused because of platelet count dose-pause rule at ≤75 × 109/L and all 3 subjects who developed platelet count ≤25 × 109/L. Subjects with ≤30% decrease in platelets from baseline were chosen as a control group (n = 30) for comparison with subjects who developed thrombocytopenia before unblinding. Data were analyzed by treatment group only after completion and unblinding of NEURO-TTR. For subjects with thrombocytopenia, samples were tested: at baseline, at a time point midway between baseline and platelet-nadir, at platelet-nadir, and in some cases, postplatelet-nadir. For subjects with no significant fall in platelets, samples were obtained at baseline and between 3 and 6 months of dosing.
Multiplex biomarker analysis of baseline plasma samples of subjects before NEURO-TTR
Multiple analyte profiles (MAP) of various human cytokines, chemokines, and growth factor concentrations were obtained from plasma samples (Supplementary Table S5) at baseline from 59 subjects on placebo and 109 subjects on inotersen (n = 168, of 172 subjects) in NEURO-TTR using a Luminex™ microsphere-based assay at Myriad-Rules-Based Medicine (Austin, TX).
Statistical analyses
The number and percent of subjects with positive findings for antiplatelet IgM (drug dependent and drug independent), antiplatelet IgG (drug dependent and drug independent), anti-PF4 IgM, anti-PF4 IgG, and platelet antibody bead array (PABA) were tabulated for NEURO-TTR, OLE, and both trials combined. Treatment emergent was defined as a negative baseline value followed by any positive postbaseline value. The correlations between change in platelet count and percent change in IgM were analyzed using Pearson correlation analysis. Association between total IgM and anti-PF4 IgM was analyzed using chi-square test or Fisher's exact test. The difference in total IgM between anti-PF4 IgM positive versus negative was assessed using a t-test. Statistical analysis was conducted by Parexel (Boston, MA).
Comparisons of MAP data between inotersen and healthy volunteers were analyzed with a paired Student's t-test. Multiple comparisons were evaluated with a one-way analysis of variance, followed by a post hoc Dunnett's test with JMP version 13 (SAS Institute). A value of P ≤ 0.05 was considered significant.
All data included were from studies receiving institutional regulatory approval (Supplementary Table S6).
Results
Thrombocytopenia in NEURO-TTR and OLE Studies
In general, platelet count reductions occurred gradually over several weeks with mean nadir typically occurring within the first 3–6 months of treatment and nadir platelet count values ≥100 × 109/L in most (∼80%) subjects in NEURO-TTR (Table 1). Subjects whose platelet counts fell <140 × 109/L were grouped under the following grades of thrombocytopenia to facilitate risk mitigation strategies, that is, institution of corticosteroids and/or discontinuation of the drug.
Confirmed Nadir Platelet Count in Subjects Participating in NEURO-TTR
A confirmed value is based on consecutive laboratory values obtained within a 7-day time period and grades are based on CTCAE values.
Modified CTCAE values to facilitate safety monitoring for dose pause and corticosteroid administration.
Source: Results are from central and local laboratory values combined and each subject is only counted once in the lowest category.
CTCAE, Common Terminology Criteria for Adverse Events; TTR, transthyretin.
This trend was also observed in ∼62% placebo-inotersen subjects in OLE (Fig. 1). In NEURO-TTR, there was a higher incidence of all grades of thrombocytopenia as compared with placebo (Table 1). In the OLE study, 54.4% of subjects overall had confirmed nadir platelet counts <140 × 109/L. In placebo-inotersen subjects, this was 47.5% (Table 2). Platelet reductions were reversible with pausing or discontinuation of dosing. No difference between treatment groups was observed in the incidence of bleeding events during either study (data not shown).
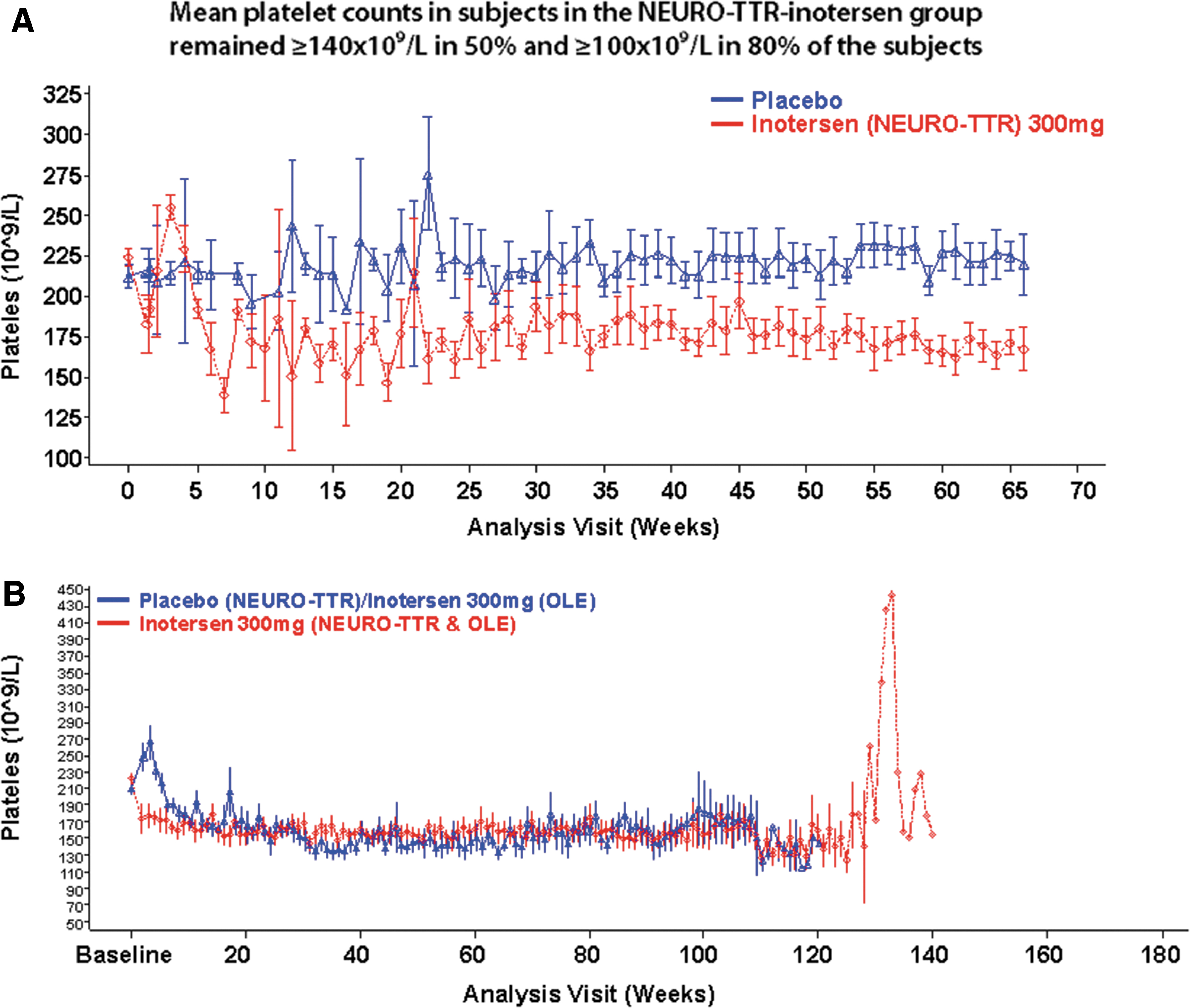
Mean (±standard error) platelet count over time for subjects enrolled in NEURO-TTR
Confirmed Nadir Platelet Count in Subjects Treated in Open-Label Extension
A confirmed value is based on consecutive laboratory values obtained within a 7-day time period and grades are based on CTCAE values.
Modified CTCAE values to facilitate safety monitoring for dose pause and corticosteroid administration.
Source: Results are from central and local laboratory values combined and each subject is only counted once in the lowest category.
Three subjects treated with inotersen in NEURO-TTR developed grade 4 thrombocytopenia (≤25 × 109/L). One grade 4 subject, IN13 (key in Supplementary Table S2), presented with intracranial hemorrhage and died before treatment could be initiated. Two other grade 4 cases recovered after treatment with corticosteroids and permanent discontinuation of inotersen. Rapid platelet count recovery in the individuals with grade 4 thrombocytopenia after institution of corticosteroids and discontinuation of the drug suggests an underlying immune-mediated mechanism. No subjects in OLE showed grade 4 thrombocytopenia.
Anti-PF4 and antiplatelet antibody estimation in NEURO-TTR and OLE
There was no significant baseline or treatment-emergent anti-PF4 IgG detected in either placebo or inotersen-treated subjects (Table 3). One subject was anti-PF4 IgG positive in NEURO-TTR-placebo and one inotersen-treated subject in OLE. These were considered incidental findings because these either occurred before dosing or were not associated with thrombocytopenia.
Antibody Status in Subjects Treated in the NEURO-TTR Clinical Trials
Patients with missing values were not included in the analysis.
Treatment emergence was defined as negative baseline value followed by any postbaseline value. Positive postbaseline values for subjects whose baseline values were missing were not considered to be treatment emergent.
OLE, open-label extension.
Incidence of treatment-emergent anti-PF4 IgM increased over baseline in inotersen-treated subjects in NEURO-TTR. Treatment-emergent incidence was 20% and 26% in inotersen-treated subjects in NEURO-TTR and OLE trials, respectively (Table 3). However, there did not appear to be an increased incidence in subjects who switched from placebo to inotersen on entry to OLE. The role for treatment-emergent anti-PF4 IgM in precipitating thrombocytopenia is unclear because anti-PF4 IgM is generally not believed to be causally related to thrombocytopenia (Supplementary Table S2). As discussed hereunder, it is possible that this increased incidence is reflective of an overall increase in total plasma IgM after inotersen treatment. In general, a HIT-like mechanism did not play a role in inotersen-mediated platelet decrease in either NEURO-TTR or OLE extension studies.
A low incidence of antiplatelet IgM-positive or IgG-positive subjects was noted at baseline in the inotersen arm of NEURO-TTR (Table 3). Five patients (IN1, IN4, IN5, IN9, and IN10) who were antiplatelet IgG positive at baseline continued to test positive during NEURO-TTR. Of these, subjects IN4 and IN5 developed grade 1b thrombocytopenia, subjects IN9 and IN10 developed grade 2 thrombocytopenia, and subject IN1 maintained normal platelet counts (Supplementary Table S2). Inotersen-dependent antiplatelet IgG-positive status in one subject at baseline (Table 3) is considered to either reflect preexisting antiplatelet antibody binding to single-stranded DNAs in general [11] or endogenous antiplatelet antibodies that are reactive for domains that overlap with the polyanionic DNA [12] in the presence of platelets [13] and therefore considered nonspecific binding to the drug in vitro.
Treatment-emergent antiplatelet IgM was detected in 3% and 33% of subjects in NEURO-TTR and OLE, respectively, and antiplatelet IgG in ∼17% and ∼78% in NEURO-TTR and OLE, respectively (Table 3). In NEURO-TTR most of these antiplatelet antibodies were determined to react with platelets in the absence of inotersen (drug-independent) antibodies.
Of the 24 subjects who became antiplatelet IgG positive after treatment in either NEURO-TTR or OLE, 18 subjects had a platelet count ≤140 × 109/L, of which 5 (20%) subjects (IN11, IN12, IN13, NTE10, and IN7E) had a platelet count ≤50 × 109/L (Supplementary Table S2). Of these, subject IN7E was also antiplatelet IgM positive during G3 thrombocytopenia.
The timing of antiplatelet IgG positivity was also highly variable among five subjects whose platelet counts fell <50 × 109/L. In subjects IN11 and IN13, the emergence of antiplatelet IgG was concomitant to the onset of severe thrombocytopenia in NEURO-TTR, whereas in subject IN12 antiplatelet IgG emerged 2 weeks before and during the onset of G4 thrombocytopenia in NEURO-TTR. Another subject, NTE10, who was antiplatelet IgG negative during NEURO-TTR, became antiplatelet IgG positive during the first week of OLE, notably during a downward trend in platelet counts from G1a to G1b thrombocytopenia. Subject IN7E who was antiplatelet IgG/IgM positive in NEURO-TTR, tested antiplatelet antibody negative in OLE for several weeks until turning antiplatelet IgG positive during G3 thrombocytopenia.
Despite strong correlation of antiplatelet IgG-positive status with various grades of thrombocytopenia in NEURO-TTR, it is notable that all three subjects IN11, IN112, and IN13 with G4 thrombocytopenia (<25 × 109/L) were antiplatelet IgG positive, of which subjects IN12 and IN13 were GPIIb/IIIa positive (Supplementary Table S2), an epitope commonly associated with immune thrombocytopenia. Reversal of thrombocytopenia by corticosteroids (the first-line treatment for immune thrombocytopenia) in patients IN11 and IN12 (data not shown), aided by discontinuation of the drug suggests an underlying immune-mediated mechanism in inotersen-induced thrombocytopenia.
Immune dysregulation in NEURO-TTR subjects
Total IgG and IgM were measured in some subjects in NEURO-TTR and OLE. Although postdose IgG did not change significantly from baseline in either inotersen-treated or placebo arm (Supplementary Fig. S2), postdose IgM increased from baseline in inotersen-treated subjects in NEURO-TTR; placebo-treated subjects had no IgM increase (Fig. 2). Based on available data, the timing of IgM increases largely occurred within the first 12 weeks after inotersen exposure, with levels maintained during continued treatment (Supplementary Tables S3 and S4). Increases in total IgM production are consistent with a polyclonal expansion, perhaps secondary to a general increase in B cell activation.

Scatter plot of IgM maximum percent change from baseline versus percent change of nadir platelet for first 6 months (on-treatment) of NEURO-TTR and OLE. Inotersen-treated set correlation coefficient = 0.1539; NEURO-TTR placebo correlation coefficient = 0.68549.
The primary objective of these investigations was to identify baseline biomarkers that would identify hATTR-PN subjects at risk for developing thrombocytopenia upon initiation of inotersen. Of more than 60 analytes measured, B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL; ligands of TNSF receptors exclusively expressed on B lymphocytes) [14] were found to be above Rules-Based-Medicine (RBM) normal reference ranges for a significant number of subjects. BAFF and APRIL were significantly (P ≤ 0.001) higher in NEURO-TTR subjects (mean ± standard deviation 1,252 ± 537 pg/mL and 9 ± 5 ng/mL, respectively) compared with samples from healthy volunteers (1,063 ± 507 pg/mL and 2.8 ± 1.3 ng/mL, respectively) and RBM range (411–915 pg/mL and 1.4–3.8 ng/mL, respectively; Fig. 3A, B and Supplementary Table S5). Matrix metalloproteinase-9 (MMP-9; known as 92 kDa type IV collagenase, 92 kDa gelatinase or gelatinase B) a class of enzymes that belong to zinc metalloproteinases family involved in degradation of extracellular matrix (ECM) [15] was significantly (P ≤ 0.001) higher in NEURO-TTR subjects (125 ± 109 ng/mL) than both healthy controls (69 ± 30 ng/mL) and RBM (≤218 ng/mL) (Fig. 3C and Supplementary Table S5).
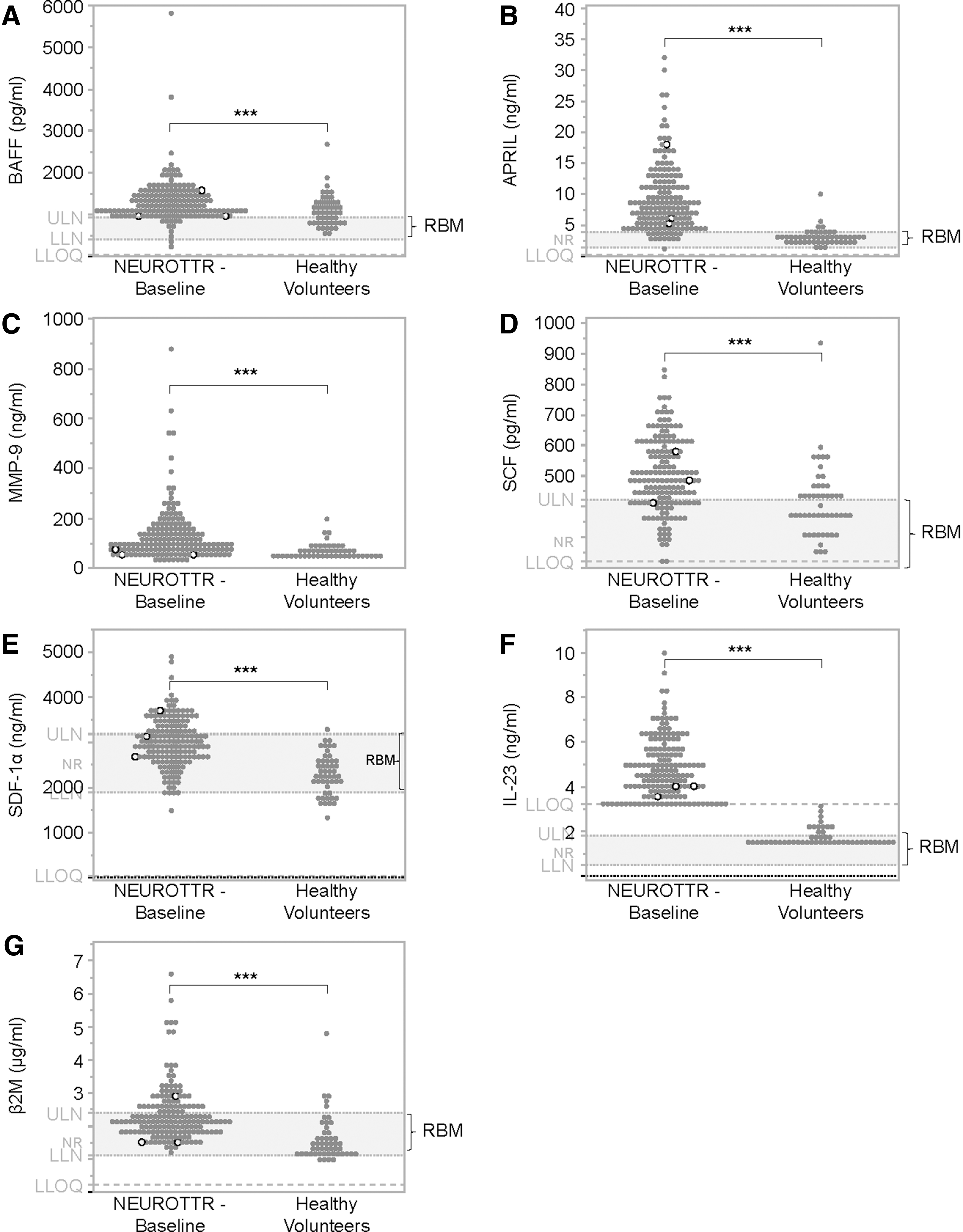
Baseline levels of
Hematopoietic and neurotrophic growth factors—stem cell factor and CXCL2, a chemokine widely expressed in the central nervous system and essential for proper functions of human neural progenitor cells (also known as stromal cell-derived factor-1, SDF-1) [16–18] were elevated significantly (P ≤ 0.001) at baseline in NEURO-TTR subjects (500 ± 120 and 2,987 ± 545 pg/mL, respectively) compared with healthy volunteers (413 ± 115 and 2,257 ± 427 pg/mL, respectively) and RBM (143–424 and 1,890–3,180 pg/mL, respectively; Fig. 3D, E and Supplementary Table S5).
Baseline levels of interleukin (IL)-23, a cytokine known to play important roles in autoimmunity and primary immune thrombocytopenia [19–22], was significantly (P ≤ 0.003) higher (4.8 ± 1.3 ng/mL) in NEURO-TTR subjects, compared with healthy volunteers and RBM range (1.7 ± 0.4 ng/mL and 0.5–1.8 ng/mL, respectively; Fig. 3F and Supplementary Table S5). Baseline level of beta-2-microglobulin (β2M), a nonspecific but relatively sensitive marker of various inflammatory and immune activation states [23], was also significantly higher (P ≤ 0.0001) in NEURO-TTR subjects (2.3 ± 0.9 μg/mL compared with healthy subjects (1.6 ± 0.7 μg/mL) and RBM range (1.1–2.4 μg/mL), respectively (Fig. 3G and Supplementary Table S5). However, there was no obvious correlation between the magnitude of the abnormality and platelet counts either in the overall population or in subjects who developed grade 4 thrombocytopenia.
Thus, IL-23 was expressed at higher levels in all NEURO-TTR subjects at baseline, compared with healthy controls, followed closely by APRIL, which is expressed at higher levels in all subjects except IN12. The three subjects with grade 4 thrombocytopenia were among subjects who had higher baseline levels of both APRIL and IL-23 (Fig. 3B, F). The presence of upregulated immune/inflammatory cytokines at baseline suggests underlying immune dysfunction in hATTR-PN subjects enrolled in the NEURO-TTR study.
Discussion
Inotersen treatment was associated with platelet count reductions, including grade 4 thrombocytopenia in three subjects (one fatal) during NEURO-TTR [8]. This occurred while platelet monitoring was carried out every 6 weeks, and before weekly testing of platelet count. Platelet reductions were mild (>100 × 109/L) in most patients, not associated with increased bleeding risk, and manageable with dose pause/regimen adjustment. Investigation of the mechanism for low platelet count excluded several important causes, including a classical HIT-type mechanism. In addition, direct bone marrow toxicity, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, thrombotic microangiopathy, sepsis, and systemic complement activation were also ruled out as contributing factors (Label for New Drug Approval Package no. 211172). Inotersen also did not activate platelets in vitro, even at concentrations >4-fold observed at Cmax of therapeutically relevant doses of ASOs (∼4.3 mg/kg in a 70-kg subject) [24], consistent with results of tests on nonhuman primate platelets upon dosing of another 2′-MOE sequence ISIS104838 [25] (Supplementary Fig. S1). Furthermore, lack of platelet activation was also observed in vitro when inotersen was incubated, at 1 μM, with various concentrations of TTR mutant proteins (data not shown).
Relevance of immune activation in gradual platelet declines and thrombocytopenia in the NEURO-TTR and NEURO-TTR extension trials
Given the polyanionic nature of ASOs, the possible role of anti-PF4 IgG was investigated. ASOs bind tightly to human purified PF4 (Kd = 8–70 nM) in vitro (data not shown). However, lack of anti-PF4 IgG response in vivo suggests that putative ASO-PF4 complexes do not meet critical physicochemical features (eg, charge density, length, and charge clustering) to create neoepitopes that are produced by unfractionated heparin binding to PF4 [9].
The likely mechanism for mild reductions in platelet count may relate to innate immune activation; for example, higher total serum IgM observed with 300 mg/wk inotersen treatment in NEURO-TTR, leading to either increased platelet pooling and/or clearance. Total IgM increases are a hallmark of oligonucleotide-mediated innate immune cell activation and B cell stimulation [25,26]. Platelet pooling may be secondary to either or both increased inflammatory activity and greater sequestration because of increased adherence to vascular endothelium [27] or because of antibody binding. Increases in total plasma IgM can increase rate of platelet clearance after inotersen treatment by facilitating direct interaction with functional IgM clearance receptors (eg, Fcα/μ receptor) and/or complement receptor [28] on macrophages thereby enhancing their phagocytic capacity [29–33]. Although platelet sequestration studies have not been conducted in human subjects treated with inotersen, this assertion is supported by 111Indium-labeled platelet studies conducted in nonhuman primates that showed increased localization of platelets to liver and/or spleen with another 2′-MOE ASO (ISIS 104838), which produced a consistent and self-limiting decrease (≤150 × 109/L) in platelets and severe thrombocytopenia [25].
A possible mechanism for the emergence of antiplatelet IgM and anti-PF4 IgM antibodies is an expansion of B lymphocytes through innate immune activation, especially B1 cells. B1 cells [34] are a relatively small but unique subpopulation of B lymphocytes that produce background levels of natural IgM antibodies. These lymphocytes are known to contain pattern recognition receptors such as TLR 9 [35–37], which can be stimulated in a concentration- and sequence-dependent manner by synthetic oligonucleotides [25,38]. Natural anti-PF4 IgM antibodies have been detected in cord blood of newborn babies, which supports the notion that these are included within the repertoire of IgM produced by these cells [39].
Severe thrombocytopenia observed in a small number of patients appears to be because of the emergence of antiplatelet IgG antibodies, detected by a flow cytometry-based immunofluorescence method, a valid test for detecting antiplatelet antibodies [40,41]. PABA tests further revealed that these antiplatelet IgG antibodies targeted GPIIb/IIa epitope. Antiplatelet IgG was observed in 29% inotersen-treated subjects tested in NEURO-TTR and 54% in OLE, yet only a fraction of them developed severe thrombocytopenia. Therefore, additional susceptibility factors may be at play including individual affinity of these IgG antibodies for various Fcγ receptors (high affinity versus low affinity) to which they may bind [42]. Of importance, these antiplatelet IgG were classified as drug independent that is consistent with them being part of the repertoire of antibodies produced by the polyclonal expansion of B cells, although class switching cannot be ruled out. Despite lack of 100% correlation of antiplatelet IgG-positive status with various grades of thrombocytopenia in NEURO-TTR, it is notable that all three subjects (IN11, IN112, and IN13) with grade 4 thrombocytopenia (<25 × 109/L) were antiplatelet IgG positive, of which two (IN12 and IN13) were GPIIb/IIIa positive (Supplementary Table S2), an epitope commonly associated with immune thrombocytopenia. Two of these subjects also tested positive for anti-PF4 IgM, but this is thought to be a nonspecific finding because of lack of known causal association between anti-PF4 IgM and thrombocytopenia.
Although ASOs can cause innate immune activation, the effect is both dose and sequence dependent. An inflammation-mediated mechanism for low platelet count in monkeys treated with these compounds has been suggested [25], and this may explain the sequence-dependent effect of 2′-MOE ASOs on platelet count described in the evaluation of the Ionis Integrated Safety Database [43].
Evidence of underlying immune dysregulation and proinflammatory profile of hATTR-PN subjects
The investigation also considered why the incidence of grade 4 thrombocytopenia differed from the experience of earlier clinical trials. Our data show evidence of an underlying immunological dysregulation in hATTR-PN subjects, characterized by higher baseline levels of cytokines involved in autoimmunity (BAFF and APRIL) and primary immune thrombocytopenia (IL-23) [14,19–22]. Higher baseline levels of chemokines such as SDF-1α/CXCL2 secreted from platelets and neurons [44–47], ECM breakdown product (MMP-9) [20], and proinflammatory marker such as β2M [23] can be downstream products of innate immune activation, through interaction with receptor for advanced glycation end product (RAGE) by proinflammatory TTR aggregates and amyloid fibrils in vivo [48–54].
Although the severity of platelet decrease did not correlate with baseline levels of BAFF, APRIL, and IL-23, subjects with grade 4 thrombocytopenia were among those who had higher baseline levels of these proinflammatory cytokines compared with healthy controls and normal range. Evidence of B cell and immune activation through T cell receptor pathways in symptomatic V30M hATTR-PN subjects gleaned from peripheral blood cell gene expression profiling further supports the role of immune activation in hATTR [55]. Collectively, increased levels of BAFF, APRIL, and IL-23 at baseline in hATTR-PN subjects in NEURO-TTR suggest immune dysregulation predisposing some subjects during inotersen treatment to immune-mediated thrombocytopenia mediated by antiplatelet IgG antibodies, particularly those targeting GPIIb/IIIa epitope. Detailed internal investigations (unpublished) revealed that a severe proinflammatory condition exists, before administration of volanesorsen in familial chylomiocronemia patients, with elevated proinflammatory cytokine profiles although different from the TNF signaling pathway observed in hATTR. Although detailed investigation of the inflammatory status associated with severe thrombocytopenia in Deschene's muscular dystrophy have not been studied in detail, it is noteworthy that the extremely high triglyceride levels or muscle degeneration that occur in these diseases, respectively, are also associated with inflammatory background [56].
TLR9 engagement by inflammatory ASOs can potentially upregulate cytidine deaminase activity, a key element of the B cell class switch-inducing machinery, followed by class switch DNA recombination from C(μ) to C(γ)1, C(γ)2, and C(γ)3 germline transcription [57]. In the context of the polyclonal B cell stimulation observed in these subjects, it is possible that class switching from antiplatelet IgM to IgG in some individuals leads to treatment-emergent antiplatelet IgG in some subjects, with epitope specificity appearing to be an important determinant of severity of platelet reduction.
In conclusion, reduced platelet counts observed in NEURO-TTR appears to be a combination of immune dysregulation in hATTR patients (eg, abnormally high levels of APRIL and IL-23) coupled with polyclonal expansion of lymphocytes in response to 300 mg/wk inotersen treatment, tipping the balance in some patients, in favor of antiplatelet IgG antibodies leading to more severe thrombocytopenia. Since most antiplatelet IgG-positive subjects did not experience severe thrombocytopenia and the timing of the positive tests with thrombocytopenia was highly variable, testing for antiplatelet IgG antibodies during treatment is unlikely to help allow early detection of subjects that are at risk of severe thrombocytopenia and frequent monitoring remains the most robust means to avoid severe thrombocytopenia. Finally, cytokine testing is also not sufficiently predictive to help identify patients at risk of significant falls in platelet count.
Footnotes
Acknowledgments
The authors thank the patients who participated in this trial and their families; employees of Ionis (Dr. Elizabeth A. Ackermann; Richard Geary, PhD, Brenda Baker, PhD, and C. Frank Bennett, PhD) and Akcea Pharmaceuticals (Dr. Michael Stevenson, RPh, PhD) for their help in conducting this study and critical review of an earlier version of the article; Christine Hoffmaster, BS, and Joseph Schroeder, MS, for technical support; Robert Saunders and Tracy Reigle for assistance with formatting; Inotersen project team; and Ushasree Narayanan, DVM, MS, of CelamzaLLC, for her assistance in proofreading of the article.
Author Disclosure Statement
S.J.S. is a consultant for Ionis Pharmaceuticals, Inc. and does not hold stock in the company. J.L.W. is a consultant for Ionis Pharmaceuticals, Inc. and holds stock in the company. B.R.C. is a consultant for Ionis Pharmaceuticals and Director, Neutrophil and Platelet Immunology, Blood Center Wisconsin, Milwaukee, WI, where antiplatelet and anti-PF4 work was performed and does not hold stock in the company. P.N., L.S., E.S., J.A.T., S.P., S.A.B., L.-J.T., T.M., J.L.W., S.X., J.A.E., S.P.H., B.M., and S.G.H. are employees of Ionis Pharmaceuticals, Inc.
Funding Information
This study was fully funded by Ionis Pharmaceutical, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.