
Abstract
Neuroblastoma (NB) is the most common solid tumor in childhood. Twenty percent of patients display MYCN amplification, which indicates a very poor prognosis. MYCN is a highly specific target for an NB tumor therapy as MYCN expression is absent or very low in most normal cells, while, as a transcription factor, it regulates many essential cell activities in tumor cells. We aim to develop a therapy for NB based on MYCN silencing by short interfering RNA (siRNA) molecules, which can silence target genes by RNA interference (RNAi), a naturally occurring method of gene silencing. It has been shown previously that MYCN silencing can induce apoptosis and differentiation in MYCN amplified NB. In this article, we have demonstrated that siRNA-mediated silencing of MYCN in MYCN-amplified NB cells induced neurogenesis in NB cells, whereas retinoic acid (RA) treatment did not. RA can differentiate NB cells and is used for treatment of residual disease after surgery or chemotherapy, but resistance can develop. In addition, MYCN siRNA treatment suppressed growth in a MYCN-amplified NB cell line more than that by RA. Our result suggests that gene therapy using RNAi targeting MYCN can be a novel therapy toward MYCN-amplified NB that have complete or partial resistance toward RA.
Introduction
Neuroblastoma (NB) is one of the most common solid tumors in early childhood, accounting for about 7% of pediatric malignancies and ∼15% of cancer-related deaths in childhood [1,2]. NB arises from primitive cells of the neural crest [3], which normally differentiate to form the nervous system and adrenal medulla [4]. Amplification of the proto-oncogene MYCN is strongly correlated with a poor prognosis in NB, and accounts for ∼20% of cases. Like other cancers, current NB therapy includes surgery, chemotherapy, monoclonal antibody treatment, and radiotherapy [5]. The NB tumors vary remarkably in their response to treatment based upon their stage and biological features [6]. Even MYCN-amplified NB may achieve remission after surgery alone if it remains localized [5]. Chemotherapy treatment with 13-cis-retinoic acid (RA) has become the standard therapy in high-risk NB, which induces neuronal differentiation [7] and maintains NB as a minimal residue disease [8]. However, the emergence of 13-cis-RA resistance eventually leads to relapse in most cases [9]. Supplementation of therapy with anti-GD2 antibody therapies combined with granulocyte macrophage colony-stimulating factor and interleukin-2 (IL-2) has improved the 2-year survival rate by 20% [7,10,11], but for patients developing resistance to RA, the prognosis of MYCN-amplified NB remains very poor [11,12].
N-Myc is a member of the Myc family and a bHLHZip transcription factor, controlling many genes involved in the regulation of essential cellular activities [13,14]. Myc proteins, including N-Myc, act as both transcriptional activators and repressors [15–17]. N-Myc also binds at, or around, the transcription start site of genes that have already been transcriptionally activated and amplifies transcription of the genes [18]. Overexpressed N-Myc induces proliferation, suppresses apoptosis and differentiation, and so promotes tumorigenesis of NB [13]. N-Myc is a transcription factor, for which it is typically very difficult to develop drugs; hence, N-Myc is a good potential target for genetic therapy by short interfering RNA (siRNA)-mediated silencing.
Studies on N-Myc expression in adult mice suggest levels that are very low or absent in the brains and other organs, although it is expressed in the forebrain, hindbrain, and in kidney of newborn mice [19]. This suggests that a MYCN-targeted siRNA therapy is unlikely to have any significant off-target effect since it is unlikely to enter the brain due to the impermeability of the blood–brain barrier. There have been several proof-of-concept studies into MYCN siRNA therapies [20–23], but none has emerged as a clinical product so far, and further studies are required into the mechanism and safety of this approach.
siRNA is a naturally occurring method of gene silencing first described in 1998 [24]. Over the last 20 years, there have been numerous attempts to develop clinical applications of siRNA and the first siRNA-based drug Patisiran (Onpattro), for transthyretin amyloidosis, was approved for clinical use by the U.S. Food and Drug Administration (FDA) in August 2018 [25]. RNA interference (RNAi) is a promising approach for cancer therapy and has been used, for example, in the knockdown of oncogenes such as neuron growth factor (NGF) in pancreatic cancer [26] and stearoyl-CoA desaturase-1 in liver cancer [27]. Several siRNAs for cancer treatment have entered clinical trials, such as Atu-027 targeting protein kinase N3 gene for therapy of advanced solid tumors, and SPC2996 targeting Bcl-2 gene for chronic lymphocytic leukemia [28]. While there is undoubted potential for siRNA cancer therapies, problems of delivery have proven difficult to overcome, but evidence with Patisiran, involving high-efficiency delivery of siRNA to the liver by lipid nanoparticles (LNPs) [25], reinforces the belief that, if the delivery issue can be solved, there is the prospect for a wide range of novel therapies for cancer.
Thus, in other areas of our research, we are developing genetic approaches to therapy of NB with novel nanoformulations based on mixtures of targeting peptides and lipids, which can be delivered systemically. For example, we have delivered plasmid DNA expressing IL-2/IL-12 cytokine adjuvant immunotherapies to tumors in the syngeneic murine model of NB, leading to tumor eradication [29,30]. We have also developed lipid/peptide nanoformulations for siRNA packaging and delivery [31–33] and aim to use this in developing novel NB therapies. In this article, we have investigated on the therapeutic potential of MYCN silencing by RNAi in MYCN-amplified NB cells in vitro, focusing on the potential for treatment of disseminated, RA-resistant tumors. We have further investigated the hypothesis that MYCN silencing by siRNA could limit proliferation and promote differentiation of NB cells by analyzing the expression of proteins involved in differentiation, in cell replication, and in the production of neurites, characteristic of differentiated neurons. The results of this study are a key step toward the development of the therapeutic potential of MYCN siRNA, which will enable us to progress to therapeutic studies in vivo.
Materials and Methods
Cell culture
Human MYCN-amplified NB cell lines Kelly and SK-N-BE(2) were cultured in RPMI1640+GlutaMAX (Thermo Fisher Scientific, Northumberland, UK) with 10% fetal bovine serum (FBS; Sigma-Aldrich, Dorset, UK), 25 mM HEPES buffer, and 100 U/mL Penicillin/Streptomycin (P/S; Thermo Fisher Scientific). Non-MYCN-amplified cell lines LAN-5 and SK-N-SH cells were cultured in Minimum Essential Medium Eagle (MEM; Sigma-Aldrich) with 10% FBS, 2 mM
Short interfering RNA
The anti-MYCN siRNA (siMYCN) and negative control siRNA (nontarget control pool) (siNeg) (both ON-TARGETplus siRNA, Dharmacon, Cambridge, UK) were used in this study. The sequence of siMYCN sense strand is 5′-CAGCAGUUGCUAAAGAAAAUU-3′, while the antisense strand was 5′-UUUUCUUUAGCAACUGCUGUU-3′.
siRNA transfections
Before transfection, NB cells were seeded at 8 × 104 cells per well in 24-well plates in complete media without P/S and incubated overnight in an incubator at 37°C in 5% CO2. The confluency reached ∼50% the next day. SiMYCN (custom)/siNeg (5–50 μM) (ON-TARGETplus siRNA; Dharmacon) was mixed with Lipofectamine RNAiMAX (RNAiMAX; Thermo Fisher Scientific) at a 1:1 volume ratio in OptiMEM (Thermo Fisher Scientific). After 10 min of incubation, the nanocomplexes were added to cells in complete culture media, and the plates were centrifuged at 400 g for 5 min immediately. The cells were incubated for 48 h at 37°C in 5% CO2.
Quantitative real time RT-PCR
Total RNA was extracted using an RNeasy mini kit (Qiagen, Manchester, UK), and then, 10–50 ng total RNA was mixed with the SensiFAST Probe Hi-ROX one-step kit (Bioline, London, UK) according to the company's instruction. TaqMan probes (Thermo Fisher Scientific) used in this study were as follows: Human ActB (Assay ID: Hs01060665_g1), Human MYCN (Hs00232074_m1), and Human NTRK1 (Hs01021011_m1). The cycles were performed in the Bio-Rad 96 PCR machine and Ct values were obtained using Bio-Rad CFX manager (Bio-Rad Laboratories, Hemel Hempstead, UK). The silencing efficiency was calculated by Delta-Delta Ct analysis.
Differentiation in SK-N-BE(2) cells
SK-N-BE(2) cells were seeded at 5 × 104 cells per well in 12-well tissue culture plates (Nunclon; Thermo Fisher Scientific) and were incubated at 37°C in 5% CO2. Cell images were taken under bright field using an Olympus IX70 microscope with a Canon DS126191 camera attached. Five images were chosen randomly from each condition, and the experiment was performed twice. The neurite length was measured using Fiji ImageJ (Supplementary Data) with extended neurites defined as projections longer than the cell itself as SK-N-BE(2) cells naturally have short neurites (arrow in untransfected control in Fig. 4a). Neurites were traced using Straight>Freehand Line function and the lengths were measured in pixels. To smooth the freehand-drawn lines and interpolate pixel coordinates, Interpolate (Edit>Selection>Interpolate) was used with interval = 10 and “smooth.” The macro commands (below) were run every time a freehand line was drawn. Cell area was measured using the polygon selections function.
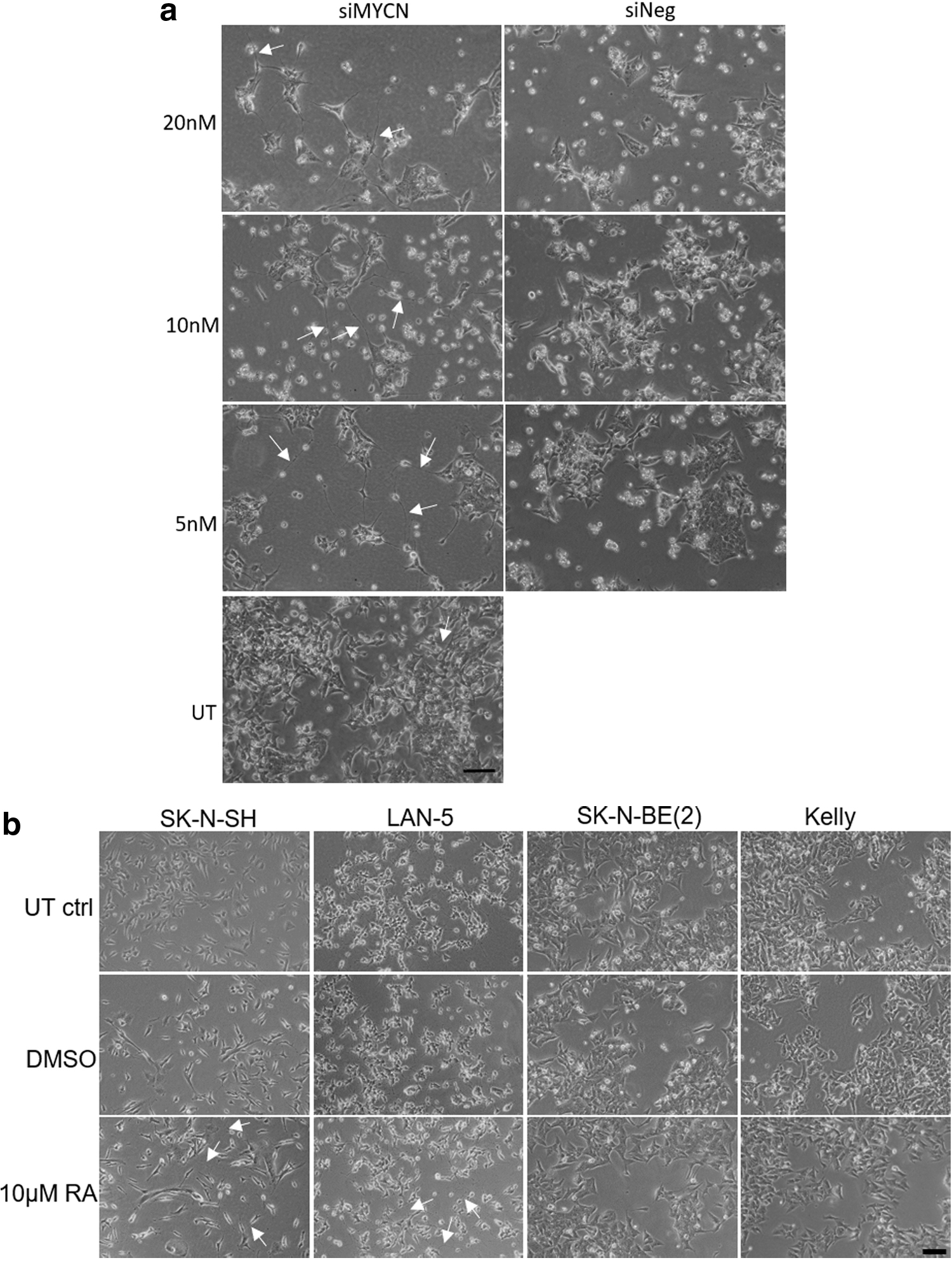
Cell morphology changes after siMYCN transfection or RA treatment.
run(“Interpolate,” “interval = 10 smooth”);
roiManager(“add”)
Proliferation assay
Resazurin method
Cells were seeded in a 96-well black plate at a concentration of 3 × 103 in 200 μL complete media per well and incubated overnight. The transfected/treated cells were incubated in an incubator under 5% CO2 at 37°C for 6 days. Twenty microliters 0.15 mg/mL Resazurin (Sigma-Aldrich) in phosphate-buffered saline was added into each well, and incubated for 3 h in an incubator. Fluorescence was measured using a 540 mm excitation/590 mm emission wavelength by a FLUOstar Optima plate reader (BMG Labtech, Aylesbury, UK).
CCK-8 method
Cells were seeded in a 96-well plate at a concentration of 6 × 103 in 200 μL of complete media per well and incubated overnight. Ten microliters Cell counting kit-8 reagent (Sigma-Aldrich) was added into each well and incubated for 3.5 h in an incubator. The absorbance at 450 nm was measured using the FLUOstar Optima plate reader (BMG Labtech).
Cell viability assay
Cells were seeded at 1.5 × 104 cells per well in 96-well plates, and were incubated in an incubator under 5% CO2 at 37°C overnight. At 24 h after transfection, the growth medium was changed and MTS reagent (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Wisconsin) was added. Cells were incubated for a further 2 h at 37°C, and then the absorbance at 492 mm was measured using the FLUOstar Optima plate reader (BMG Labtech).
Immunoblotting
The total protein of transfected cells was extracted with NP40 cell extraction buffer and the concentrations of protein in lysates were quantified using a bicinchoninic acid kit (Thermo Fisher Scientific). Up to 10 μg of total protein was mixed with 4 × loading dye buffer supplemented with 10 × DDT. It was boiled at 100°C for 5 min and then electrophoresed on 4%–12% Bis-Tris Nupage gel (Thermo Fisher Scientific) in MOPS buffer (Thermo Fisher Scientific) at 150 V for 1 h. The protein bands were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Watford, UK) in transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol) using Bio-Rad Mini Trans-Blot tank (Bio-Rad Laboratories) at 100 V for 1.5 h. PVDF membranes were then blocked with 10% dried milk in TBST (50 mM Tris-base pH 7.5, 150 mM NaCl, and 0.2% Tween-20) for 1 h and then probed and incubated with anti-MYCN (B8.4.B, 1:8,000; Santa Cruz, CA) or anti-pan Trk (B-3, 1:2,000; Santa Cruz) antibodies in 10% dried milk TBST overnight, while anti-β-Actin antibody (1:10,000; Sigma-Aldrich) was incubated for 1 h. The membranes were then washed thrice with TBST and incubated with horseradish peroxidase-conjugated secondary antibody (1:10,000; Dako, Glostrup Municipality, Denmark) in 10% dried milk TBST buffer. They were then washed with TBST again, and the protein bands were detected using the ECL chemiluminescence-based detection kit (Bio-Rad, CA), and visualized in the UV Chemi chemiluminescence detection apparatus (UV Chemistry Co., CA). The experiments were repeated thrice in SK-N-BE(2) cells transfected with siRNA and twice in Kelly cells transfected with siRNA. SK-N-BE(2) and Kelly cells treated with RA were repeated twice.
Statistical analysis
The error bars indicate the mean ± standard deviation and results were analyzed using a two-tailed, unpaired Student's t-test unless mentioned otherwise, in which case, a two-way ANOVA was performed. Probability values P < 0.05 were indicated as *, P < 0.01 were indicated as **, and P < 0.001 were indicated as ***.
Results
Cytotoxicity assay
We first investigated the potential toxicity of transfections with MYCN mRNA (siMYCN) and scrambled siRNA (siNeg) using RNAiMAX at 50 nM and 10 nM in SK-N-BE(2) cells (Fig. 1). There was no apparent toxicity of 10 nM siMYCN or 10 nM siNeg compared to untransfected control cells (P = 0.9 and 0.2, respectively), while 50 nM siMYCN and 50 nM siNeg displayed significant toxicity (P < 0.001), with only 36% and 42% of cells remaining alive. Thus, siRNA concentrations in RNAiMAX formulations of 20, 10, and 5 nM were chosen for the conditions in this study.
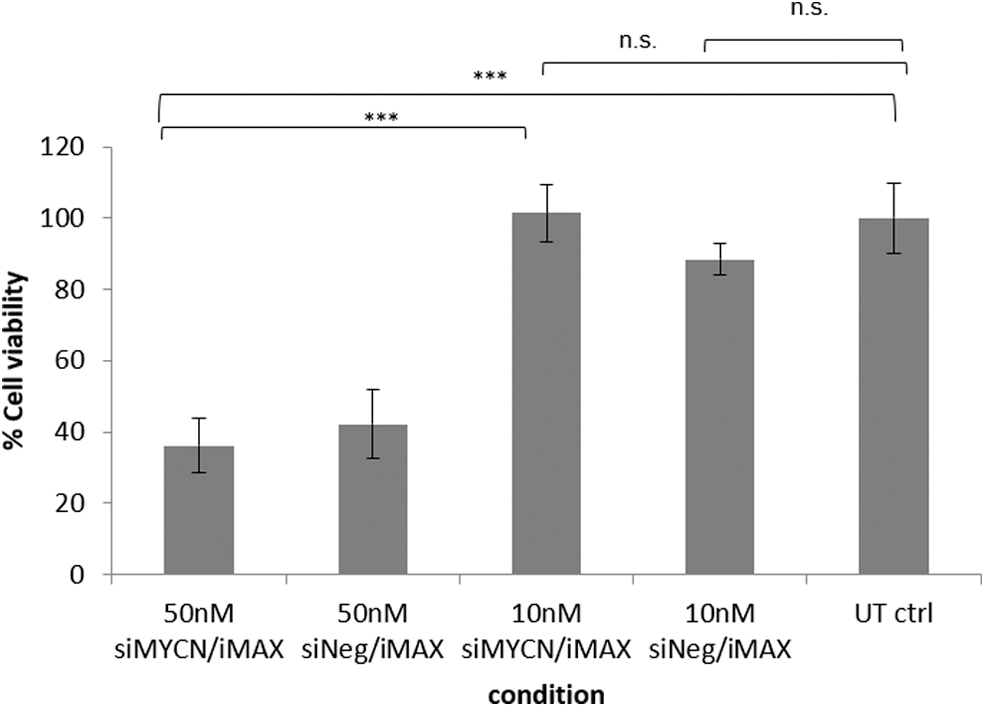
Cytotoxicity assay. Cell viability of SK-N-BE(2) cells treated with 50 or 10 nM siMYCN/siNeg was assessed using MTS assay reagent. SiRNA transfections at 50 nM using RNAiMAX were notably toxic, while the cell viability of the cells treated with 10 nM siRNAs was almost the same or slightly lower (statistically nonsignificant) to untransfected negative control cells (P < 0.001 between 50 nM siMYCN and 10 nM siMYCN, and P < 0.001 between 50 nM siMYCN and untransfected control cells). In addition, the values between 50 nM siNeg and untransfected control cells were also statistically different, with P < 0.01 indicating that toxicity was due to the formulation itself rather than MYCN silencing. The intensity was normalized to untransfected negative control cells (n = 5). In the graph, each bar represents the mean ± SD, ***P < 0.001. SD, standard deviation; siRNA, short interfering RNA.
MYCN silencing by siRNA in NB cell lines
siMYCN was chosen from three candidate siRNAs targeting MYCN mRNA (Supplementary Fig. S1a, b). MYCN mRNA/N-Myc expression levels in three NB cell lines SK-N-BE(2), Kelly, and SK-N-SH were investigated using quantitative real-time RT-PCR (qRT-PCR) and immunoblotting, respectively. The MYCN mRNA and N-Myc expression levels were elevated at 500–2,000-, and 4- to 20-fold, respectively, in the MYCN-amplified NB cell lines SK-N-BE(2) and Kelly (Supplementary Fig. S2a, b). Transfections of siMYCN (5–20 nM) using RNAiMAX were performed in the NB cell lines and MYCN silencing efficiencies were determined among the different cell lines by qRT-PCR (Fig. 2a, n = 3). In SK-N-BE(2) cells, the maximal silencing of 38% relative to siNeg was achieved at 20 nM siMYCN with statistical difference between siMYCN and siNeg also observed at 10 nM (P < 0.05), but not at the 5 nM dose. In Kelly cells, the maximal silencing was also achieved with 20 nM siMYCN (51%), relative to siNeg control (P < 0.05). In SK-N-SH cells, 20 nM, 10 nM, and 5 nM doses of siMYCN achieved similar silencing efficiencies of ∼55%, although 5 nM siMYCN silencing was not significant compared to 5 nM siNeg (P = 0.07), while those at 20 and 10 nM were (P < 0.01 and 0.001, respectively).

Relative expression of MYCN mRNA and NTRK1 mRNA in NB cell lines 48 h after siMYCN transfection quantified by qRT-PCR.
Expression of NTRK1, which encodes TrkA, a neuronal differentiation marker, was quantified by qRT-PCR in SK-N-BE(2) and Kelly cells 48 h after siMYCN transfections (Fig. 2b) to assess the induction of a downstream differentiation marker after MYCN silencing. TrkA receptor is expressed on neuronal cells and binds NGF with high affinity inducing differentiation. TrkA expression is upregulated by N-Myc downregulation [13,34,35]. In SK-N-BE(2) cells, NTRK1 was upregulated by siMYCN-mediated MYCN silencing by 2.0- to 3.3-fold compared to siNeg (Fig. 2b) (P < 0.05 for all concentrations), while in Kelly cells, NTRK1 was upregulated 1.3- to 4.4-fold over siNeg-treated cells (P < 0.05 for each concentration) (Fig. 2b, n = 3).
Comparison of MYCN silencing and Trk upregulation by siMYCN and RA at the protein level
We then investigated the effects of siMYCN-mediated MYCN silencing on protein expression by western blot analysis. SK-N-BE(2) and Kelly cells were transfected with siMYCN or siNeg at 20, 10, and 5 nM and were harvested 72 h after transfection. Western blots were probed with antibodies to N-Myc, pan-Trk, which stains for TrkA, B, and C, and β-actin as a house keeping gene (Fig. 3a, b). The dose–response of N-Myc and Trk to all-trans RA of SK-N-BE(2) and Kelly cell lines was also assessed in a range of concentrations up to 25 μM. In previous studies, NB cells were treated with RA in vitro up to 10 μM [36,37], while the 5–10 μM RA doses are equivalent to the those used in clinical trials by oral administration to NB patients [38,39], and so, RA doses in this concentration range were used. The intensity of all bands from staining for N-Myc, pan-Trk, and β-actin was measured using Image J, and the values of N-Myc and pan-Trk were normalized to β-actin and to the untransfected control cells (untransfected control = 1). The relative expression level of each protein is shown under the respective band (Fig. 3). The experiments were repeated thrice or twice and the average values of the normalized N-Myc and Trk are shown (Fig. 3).
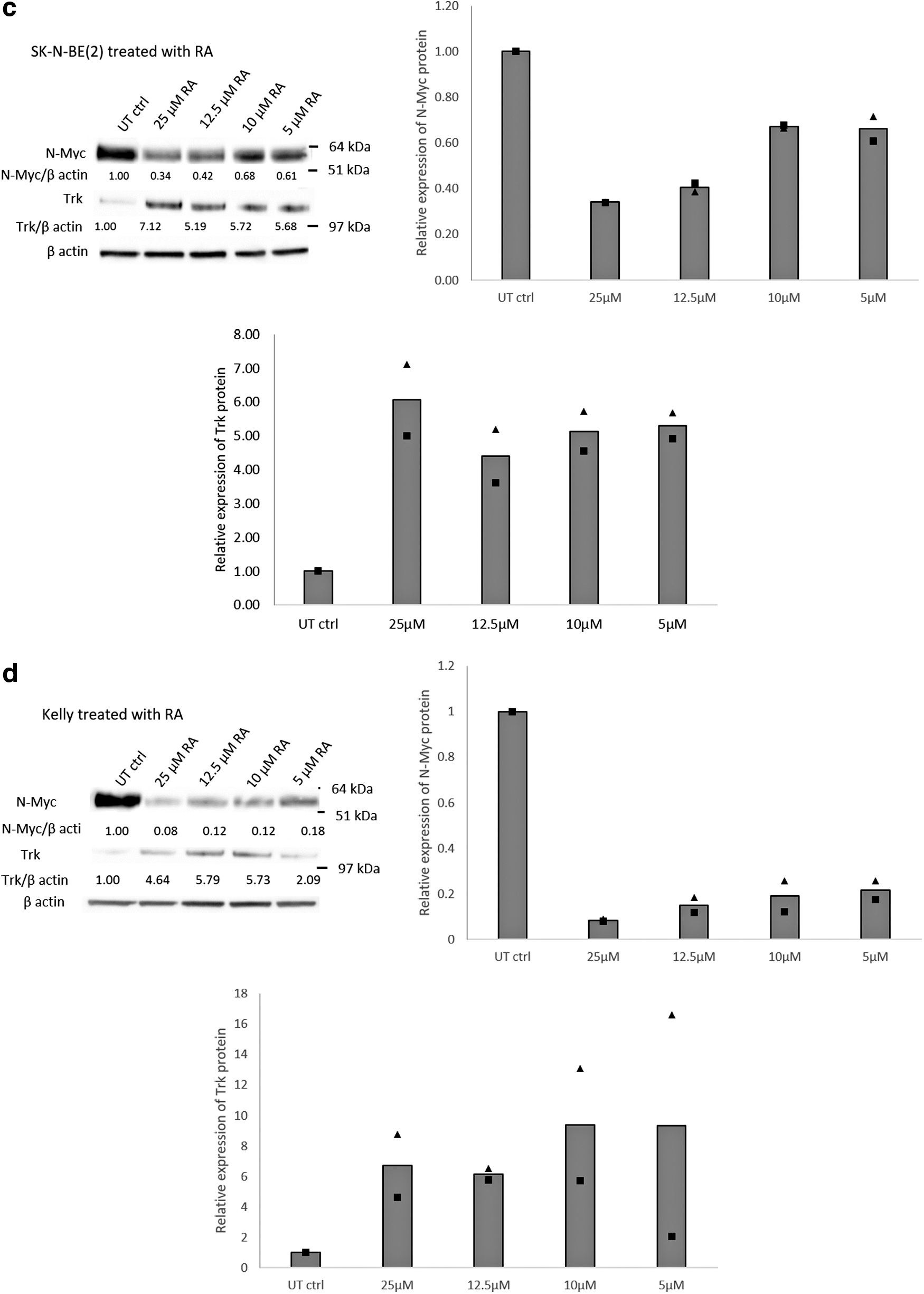
Immunoblotting of N-Myc and Trk following siMYCN transfections. The number under the band is the relative expression level calculated from the intensity of the band.
siMYCN reduced N-Myc by up to 95% in SK-N-BE(2) and up to 84% in Kelly cells (Fig. 3a, b). Furthermore, siMYCN-mediated N-Myc silencing upregulated the pan-Trk expression level by up to 2.4-fold in SK-N-BE(2) cells and 6.7-fold in Kelly cells, compared with the respective untransfected cells, while siNeg had no significant effects on protein expression. SiRNA-mediated N-Myc reduction was consistent among the three repeated experiments of SK-N-BE(2) and between the twice repeated experiments with Kelly cells. The 5 μM, 10 μM, and 25 μM RA reduced N-Myc levels by 39%, 32%, and 66%, respectively in SK-N-BE(2) cells, while N-myc was reduced by 82% in Kelly cells with 5 μM RA, with a slightly improved reduction of 92% with 25 μM RA (Fig. 3c, d). Furthermore, RA upregulated Trk expression levels in both SK-N-BE(2) and Kelly (Fig. 3c, d) cells.
Thus, N-Myc protein with 25 μM RA (66%), was equivalent to that of 5 nM siMYCN (73%) in SK-N-BE(2) cells, while in Kelly cells, N-Myc protein reduction with 10 μM RA (88%) was similar to that of 20 nM siMYCN (84%). Twenty-five micromolars RA achieved the highest Trk expression level in SK-N-BE(2) cells (7-fold increase), while siMYCN did not achieve this level with a maximum of 2.3-fold increase of Trk. Thr 12.5 μM RA treatment of Kelly cells led to a 5.8-fold increase in Trk similar to 5nM siMYCN (6.5-fold increase).
In SK-N-BE(2) cells, N-myc and Trk levels were fairly constant at all three doses of siMYCN. Kelly cells showed a shallow dose–response to siMYCN of reducing N-Myc, but this did not correlate with a Trk dose–response. Only with Kelly cells was there an apparent correlation of reduced N-myc with elevated Trk in response to an increasing RA concentration.
Differentiation of SK-N-BE(2) cells by siMYCN
We then quantified neurite elongation as an index of differentiation after treatment with siMYCN or RA [8], determining the length and number of the neurites. SK-N-BE(2) transfection with siMYCN (5, 10, and 20 nM) induced significant neurite elongation with extended, interconnecting neurites, while cells also became elongated and thinner in appearance, all characteristics of differentiated cells (Fig. 4a). SK-N-BE(2) cells tend to grow in clumps, making counting of cells problematic and so, to quantify neurites, the total neurite length value per image was normalized to the total cell area rather than cell number. The experiment was performed twice and five images were analyzed in each experiment.
The neurite extensions appeared from 48 h after transfection, with a total neurite length induced by siMYCN of ∼0.015–0.025 μm/μm2 cell area (Fig. 5a). The length of neurites transfected with 20, 10, and 5 nM siMYCN on day 6 was increased from 48 h by around 0.03 μm/μm2 cell area, and each was significantly different from siNeg controls at the same concentration (P < 0.001). The increased neurite length was almost the same at the three different concentrations of siMYCN (0.033–0.035 μm/μm2 cell area) (Fig. 5a). However, although their length increased, the number of neurites did not change between days 2 and 6 (Fig. 5b). Transfections with siNeg had no effect on neurite length or number on day 2 and 6. These results suggest that siMYCN treatment induces differentiation in SK-N-B-(BE2) NB cells.
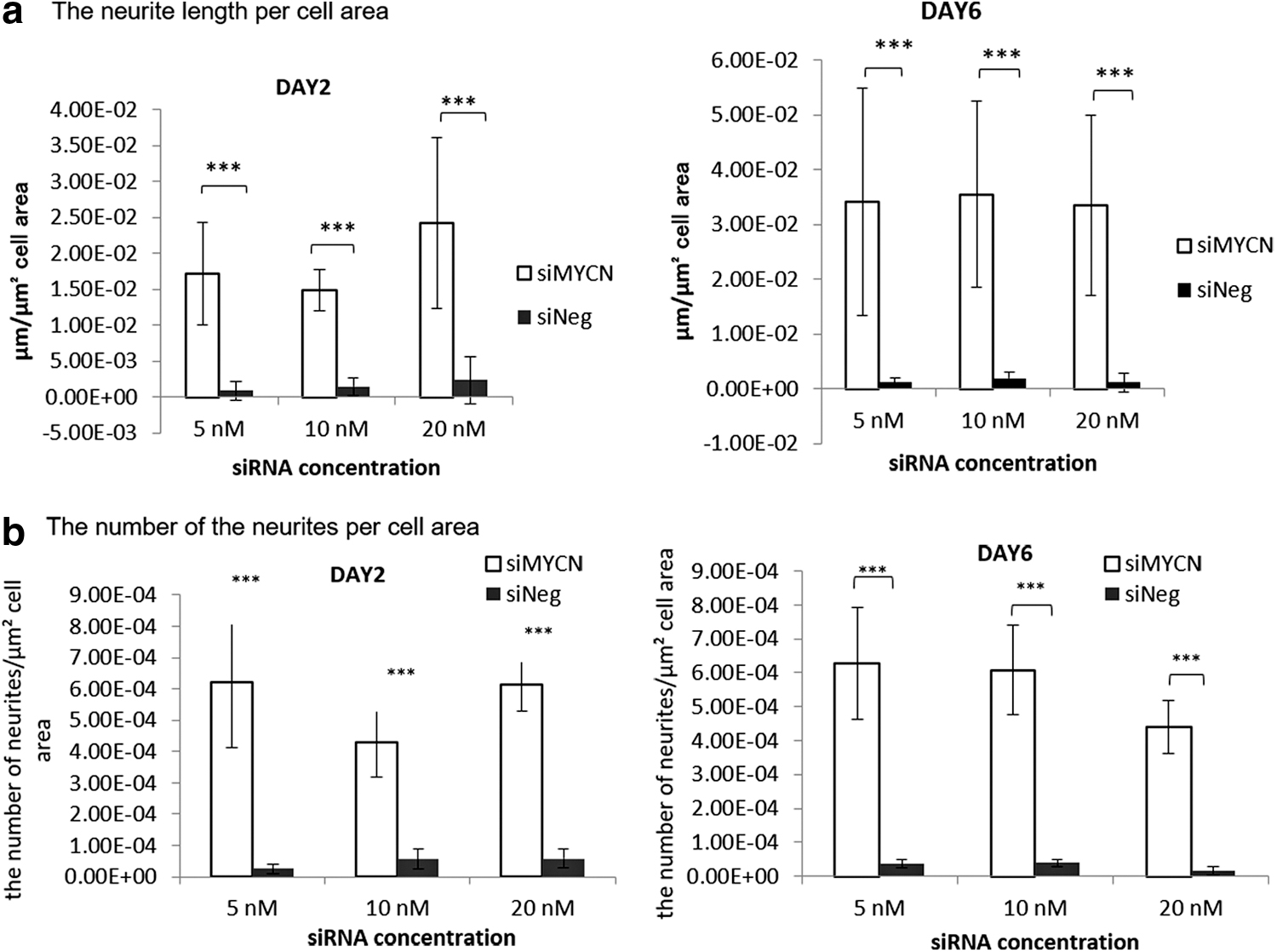
Quantification of neurites after siMYCN transfection of SK-N-BE(2) cells. siMYCN significantly induced differentiation in SK-N-BE(2) cells (n = 10), and there were significant differences in
Four different cell lines: SK-N-BE(2), Kelly, and LAN-5, which have MYCN amplification, and SK-N-SH, which is not MYCN amplified, were then treated with 10 μM RA (Fig. 4b). In SK-N-SH cell cultures, where there are normally two types of cells, neuronal and Schwann cell, the morphology of the Schwann-type cells became flatter after the RA treatment (Fig. 4b), while extended neurites formed networks and connected cells. In LAN-5, the extended neurites also formed networks among the cells after treatment with RA. On the other hand, SK-N-BE(2) and Kelly cells did not change their appearance significantly and neurite networks were not induced. Therefore, these results suggest that, despite reduced N-Myc and elevated Trk protein, Kelly and SK-N-BE(2) cells are resistant to treatment with RA.
Suppression of cell viability by siMYCN
We next compared the potential of siMYCN and RA treatment to suppress cellular proliferation. Transfection with 10 nM siMYCN suppressed cell viability over the 3 days, and the cell growth rate was relative to siNeg and untransfected control cells (Fig 6a, all P < 0.05). In addition, the cell growth rate of SK-N-BE(2) treated with 20 nM siMYCN reduced the viability, but no significant differences were observed between 5 nM siMYCN and siNeg (Supplementary Fig. S3).
We then assessed whether there was a beneficial effect of co-treatment of siMYCN and RA. The growth rate was measured using a resazurin assay at 6 days after siMYCN transfection or RA treatment (n = 3) (Fig. 6b and Table 1). The experiment was performed four times and the values were normalized to untransfected cells. Cell growth rates showed no benefit of the combined siMYCN and RA treatment, while RA on its own was also ineffective at retarding cell proliferation. However, all formulations with siMCYN were effective under all conditions (P < 0.05). These results confirm that siMYCN suppresses proliferation of SK-N-BE(2) and, here, show that it does this with much greater efficacy than RA, and so could be therapeutically beneficial in suppressing cell growth, in addition to differentiation in RA-resistant cells.

Analysis of cell viability after siMYCN treatment in SK-N-BE(2) cells.
The Statistics of the Resazurin Proliferation Assay Fig. 6b
Two-way ANOVA Bonferroni's multiple comparisons test was performed, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DMSO, dimethyl sulfoxide; RA, retinoic acid; siRNa, short interfering RNA.
Discussion
SiRNA therapies, after many years of development [24], have finally begun to realize their potential in developing therapies with the first approved and effective product, Onpattro, an LNP siRNA therapy delivered to the liver to treat transthyretin amyloidosis [25]. Delivery to tumors has been investigated extensively, including clinical trials, but so far without an approved product [26–28], but the success of Onpattro has provided fresh stimulus to the siRNA field. MYCN-targeted therapies have also been under investigation for more than 20 years, motivated by its contribution to NB tumorigenicity, particularly in MYCN-amplified tumors. As a transcription factor, MYCN is a difficult target for drug development, making siRNA an attractive approach. To develop siRNA MYCN therapies, it is important to understand the functionality of these formulations.
In this article, we have investigated the potential of MYCN silencing by RNAi as a therapy for NB by building on previous studies and further analyzing its effects on cell differentiation and proliferation. In particular, we have compared the efficacy of siMCYN in cell resistant to RA, an established treatment for NB but for which resistance can become a problem [40]. Overexpressed N-Myc induces proliferation, which is one of the characteristics of tumorigenesis in MYCN-amplified NB, and also suppresses differentiation initiated by NGF [13]. N-Myc downregulates TrkA expression by forming a repression complex with the transcription factors SP1 and MIZ1 at the core promoters of TrkA, and the complex then recruits the histone deacetylase HDAC1 to suppress transcription [41]. TrkA receptor, activated by NGF ligand, stimulates differentiation in normal cells in vitro [42], while lack of TrkA expression from overexpressed N-Myc correlates to poor prognosis in NB [43].
There is increasing evidence that MYCN is a suitable target for NBN therapies. Recent studies have shown that overexpression of N-Myc inhibits TGFB1, which is activated by RA and a ligand of transforming growth factor beta (TGF-β) signaling [44]. In addition, overexpression of N-Myc inhibits TGFB1, which is required as a differentiation modulator [44]. The Wnt/β-catenin signaling pathway is crucial for cell proliferation, differentiation, apoptosis, polarity, and pluripotency, and is thus a target for NB [45,46]. N-Myc suppression downregulates Wnt/β-catenin signaling [46], while activation of Wnt/β-catenin signaling by the transcription factor HIF1/2α leads to an undifferentiated state in NB [47]. Thus, HIF inhibition induces differentiation in NB cells by RA, by blocking the Wnt/β-catenin pathway [47]. Similarly, the combination of Wnt/β-catenin signaling inhibition and RA treatment significantly reduces NB cell viability and forces surviving cells to differentiate [48].
RA targets the RA receptor/retinoic X receptor in normal neuronal cells, and induces TrkA upregulation and differentiation in both MYCN-amplified and non-MYCN-amplified tumor cells [13], and so RA has been used as a therapy for high-risk NB for controlling minimal residue after high-dose chemotherapy [37]. However, some NB cells develop resistance to RA, such as the NB cell line SK-N-BE(2), which may result from defective retinoid signaling downstream of TrkA [49]. RA catabolism by members of the cytochrome P450 family, such as CYP26 enzymes, which specifically inhibit RA, may be involved in resistance in NB [36,50,51].
A large number of markers have been identified associated with NB differentiation [8], and in this study, we found that TrkA was particularly useful to monitor differentiation of SK-N-BE(2) cells after siMYCN treatment. TrkA mRNA expression was shown to be upregulated, while Trk proteins were shown to be upregulated by a panTrk antibody that also recognizes TrkB and Trk C, as well as TrkA. This antibody worked particularly well and along with the mRNA data provides strong evidence of upregulation of Trk differentiation markers, although it would be interesting to dissect the upregulation of each Trk in future studies. We have shown that siRNA-mediated MYCN silencing upregulates expression of Trk at both the mRNA and protein levels in SK-N-BE(2), inducing extensive neurite elongation in MYCN-amplified, p53-deficient SK-N-BE(2) cells, and suppressing cellular proliferation. In comparison, treatment of SK-N-BE(2) with 10 μM RA, in this study, reduced N-Myc by 30%, similar to previous reports [49]. This level of N-Myc reduction was similar to that with siMYCN and led to upregulated Trk expression, but no neurites or other significant differentiation-related, morphological changes were seen, suggesting that these cells are resistant to RA treatment [49]. In contrast, RA treatment in SK-N-SH and LAN-5 cells induced neurite formation and morphological changes, indicating this treatment was effective in these NB cell types. RA treatment in Kelly cells induced N-Myc silencing and enhanced Trk expression, but otherwise had no effect on the morphology.
SK-N-BE(2) cells are resistant toward not only RA but also other anticancer drugs such as doxorubicin, etoposide, cisplatin, carboplatin, and melphalan, which depend on the p53 pathway as this cell line was established from cells isolated from a patient after several sessions of chemotherapy and radiotherapy [52,53]. Our data suggest that siMYCN might be a promising therapy in cases of NB with resistance toward RA and other drugs that rely on the p53 pathway to induce apoptosis or differentiation, as observed with SK-N-BE(2) cells.
In this study, in SK-N-BE(2) cells, silencing of MYCN was siRNA dose dependent and this led to a dose-related increase of NTRK1 mRNA, although this did not lead to elevated Trk protein expression, for example, 5 nM siMYCN led to a similar level of Trk elevation as 20 nM siMYCN. Likewise, there was no difference in the induction of neurite formation from 5 to 20 nM siMYCN, indicating that even levels of MYCN silencing of 25%–30% is sufficient to induce differentiation and reduced cell growth.
However, it is important to be aware that not all NB cells respond to N-myc reduction by differentiating. In fact, MYCN expression is required for the differentiation of some NB cells [54,55]. Thus, not all MYCN-amplified NB will differentiate on MYCN suppression, due to the heterogeneity of NB.
In conclusion, we have demonstrated that siRNA mediated silencing of MYCN expression at the mRNA and protein levels, and it induces TRK1/TrkA upregulation in SK-N-BE(2) and Kelly cells. Importantly, siMYCN transfections significantly trigger neurite elongation, a differentiation marker, on SK-N-BE(2), which has nonfunctional p53 and has resistance toward RA and drug depending p53 pathway. Therefore, MYCN silencing by siRNA may provide a novel therapy for NB with drug resistance. These results will enable us to proceed to in vivo studies in murine models of NB, for which we have developed novel siRNA nanoparticle formulations [31,32].
Footnotes
Acknowledgment
We are grateful for the financial support by Dr. Michihiro Maeshima, MD.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College, London.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.