
Abstract
A safe and effective delivery system is considered a key to the success of nucleic acid therapeutics. It has been reported that pulmonary surfactants or their components could facilitate the uptake of small interfering RNA (siRNA) into the lung epithelial cells. Previously, our group investigated the use of KL4 peptide, a synthetic cationic peptide that simulates the structural properties of surfactant protein B (SP-B), as siRNA delivery vector. Although KL4 peptide exhibits good in vitro siRNA transfection efficiency on lung epithelial cells, its therapeutic potential is limited by its poor aqueous solubility due to the presence of a high proportion of hydrophobic leucine residues. In this study, we aim to address the solubility issue, designing five different modified peptides by replacing the hydrophobic leucine with alanine or valine, and assess their potential as siRNA delivery vectors. While the modified peptides retain the overall cationic property, their siRNA binding is also affected and their transfection efficiency is inferior to the parent KL4 peptide. A closer examination of the conformation of these peptides by circular dichroism shows that substitution of leucine residues leads to the change of the secondary structure from α-helical content to either β-sheet or more disordered, β-turn conformations. Relatively conservative amino acid substitutions, in terms of hydrophobicity bulk, lead to substantial conformational alteration, heavily impacting siRNA binding and release, cellular uptake, and transfection efficiency. Although the peptide modification strategy employed in this study was unsuccessful in developing an improved version of KL4 peptide for siRNA delivery, it highlights the importance of the α-helical conformation for efficient siRNA transfection, providing useful insights for future development of peptide-based RNA delivery system.
Introduction
Small interfering RNA (siRNA) has been routinely used in molecular biology as a tool for silencing specific gene through RNA interference (RNAi) mechanism. With the recent FDA approval of two siRNA drugs, namely patisiran (Onpattro™) and givosiran (Givlaari™), the application of siRNA has successfully moved from bench to bedside, marking an important milestone for siRNA therapeutics [1–3]. These two formulations use nanoparticles to deliver siRNA to the liver by parenteral injection for the treatment of hepatic diseases. To further expand the therapeutic potential of siRNA, it is necessary to explore delivery systems that can target siRNA to other organs and tissues.
siRNA is a promising candidate for the treatment of various lung diseases, including chronic obstructive pulmonary disease, respiratory infections, and cystic fibrosis [4,5]. Delivery of siRNA to the lung through topical administration allows high local concentration to be achieved with minimal systemic side effects [6,7]. With the intrinsic properties of siRNA such as large molecular size, high hydrophilicity, negative charge, and susceptibility to nuclease degradation, a transfection agent is required to promote cellular uptake of siRNA and protect siRNA from premature degradation to initiate RNAi in the cytoplasm. Interestingly, a gene silencing effect is often observed in the lungs following the delivery of naked siRNA to airways without any transfection agent [8–11], suggesting that airway lining fluids may play a critical role in facilitating siRNA uptake in the lung tissues. It is anticipated that the cationic surfactant proteins may facilitate the transfection of siRNA in the airways [12,13]. Yet, an effective delivery vector is still needed to produce robust and reliable transfection for therapeutic effect.
Cell-penetrating peptide (CPP) has been investigated for gene delivery for over two decades [14,15]. CPPs are cationic or amphipathic peptides that are developed to deliver large molecules, including nucleic acids, into cells by promoting cellular uptake and endosomal escape. Early generation CPPs are derived from natural viral proteins, signal peptides, and antimicrobial peptides. They are relatively short with insufficient charge density to be used as delivery vectors on their own, hence they are usually incorporated into existing delivery vectors such as polymers and lipids. There has been an increasing interest in designing synthetic peptides that possess the structural characteristics of CPP with the ability to function as delivery vectors alone [16].
The mechanism of CPPs to transport their cargos across cell membrane is still a subject of debate. A number of cellular uptake mechanisms have been proposed, including direct penetration across the cell membrane (eg, carpet, inverted micelle, and toroidal), endocytic pathways (eg, clathrin-mediated endocytosis and caveolae-mediated endocytosis), as well as the combination of various pathways [17]. The cellular uptake mechanism of CPP is heavily governed by its primary sequence, secondary structure as well as the cell type and cargo property. To understand this complex relationship, it is important to examine the structural characteristics when designing a peptide-based delivery system.
To this end, we investigated the use of synthetic KL4 peptide, a mimic of lung surfactant protein B (SP-B), as a potential nonviral vector for siRNA delivery. KL4 is one of the active components in lucinactant (Surfaxin), an artificial pulmonary surfactant indicated clinically for the prevention of respiratory distress syndrome in premature infants [18,19]. Our previous study demonstrated that the cationic KL4 peptide was able to form nanosized complexes with siRNA through electrostatic interaction and was successful in mediating efficient siRNA transfection in human lung epithelial cells, including A549 cells (human adenocarcinoma alveolar epithelial cells) and BEAS-2B cells (human bronchial epithelial cells). In addition, the transfection efficiency remained robust in the presence of lung surfactant, making it a good candidate for pulmonary siRNA delivery [20]. However, one of the problems associated with KL4 peptide is its poor aqueous solubility due to the high content of hydrophobic leucine in the sequences, limiting its potential for clinical application. To dissolve KL4 peptide, organic solvent such as dimethyl sulfoxide (DMSO) and acetonitrile is required, rendering it undesirable for use in pharmaceutical formulation, especially when it is designed for pulmonary delivery, due to safety concerns.
To address the solubility problem, we proposed to modify the sequence of KL4 peptide in an attempt to reduce its hydrophobicity, while preserving its transfection efficiency. KL4 is a 21-residue peptide containing hydrophobic leucine interspersed with cationic lysine (Table 1). To retain the siRNA binding ability, the number and position of lysine residues remain unchanged so that the overall net charge of the modified peptides stay positive at physiological pH. On the other hand, leucine contains an isobutyl group as side chain, making it a hydrophobic amino acid. To reduce the overall hydrophobicity of the peptide, leucine residues were either partially or completely substituted with the less hydrophobic alanine, which contains a methyl group as side chain. In addition, the replacement of leucine with valine, which contains a propyl group as side chain, was also explored, thus retaining the overall hydrophobicity of KL4, but altering its steric properties and preference for more extended conformations. The siRNA binding affinity and the structural characteristics of the modified analogs were examined. The transfection efficiency, cellular uptake, and the cytotoxicity of these analogs were studied on human epithelial cells and compared with the parent KL4 peptide.
Sequence, Average Hydrophobicity, and Molecular Weight of Peptides Used in This Study
The amino acid residues that are modified form the parent KL4 peptide are underlined. Average hydrophobicity is calculated according to the Eisenberg Consensus scale.
A, alanine; K, lysine; L, leucine; V, valine.
Materials and Methods
Materials
All the peptides used in this study (Table 1) were purchased from ChinaPeptides (Shanghai, China) with >80% purity. The peptide stock solutions were prepared at 1 mg/mL with 1% (v/v) DMSO. GAPDH siRNA, Dulbecco's modified Eagle's medium (DMEM), OptiMEM I reduced serum medium, fetal bovine serum (FBS), Antibiotic-Antimycotic (100 × ), and Lipofectamine 2000, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) were purchased from ThermoFisher Scientific (Waltham MA). Heparin (50 mg/mL) was purchased from LEO Pharma (Ballerup, Denmark). Fluorescently labeled siRNA (siGLO cyclophilin B control siRNA) was purchased from Dharmacon (Lafayette, CO). GelRed nucleic acid stain was purchased from Biotum (Hayward, CA). Anti-GAPDH antibody and anti- β-actin antibody were purchased from Abcam (Amersham, United Kingdom). Secondary antibody and Amersham ECL western blotting detection reagents were purchased from GE Healthcare (Amersham, United Kingdom). Other reagents were obtained from Sigma-Aldrich (Saint Louis, MO) as analytical grade or better.
Agarose gel retardation assay
The gel retardation assay was performed to examine the siRNA binding affinity of the peptides. Peptide/siRNA complexes were prepared at 2:1, 5:1, 10:1, 15:1, 20:1. 25:1, and 30:1 peptide to siRNA ratios (w/w), with 0.2 μg of siRNA in 10 μL of TAE buffer. Naked siRNA was included as control. The complexes were incubated for 30 min, followed by the addition of 2 μL of gel loading dye. The complexes were loaded into a 2% (w/v) agarose gel stained with GelRed. Electrophoresis was run in TAE buffer at 125 V for 25 min. The gel was visualized under the ultraviolet (UV) illumination. The release of siRNA from the complexes was also examined by using negatively charged heparin to dissociate the complexes. The peptide/siRNA complexes were formed at ratio 20:1 (w/w) and incubated for 30 min at room temperature. Heparin was added to the complexes at various heparin to siRNA ratios from 2:1 to 20:1 (w/w). The mixtures were loaded into 2% agarose gel after incubating for another 30 min and the electrophoresis was run as mentioned above.
Particle size distribution
Peptide/siRNA complexes were prepared at the ratio 20:1 (w/w) with 4 μg of siRNA in 100 μL of ultrapure water. At 30 min after complex formation, the hydrodynamic size of the complexes was measured by dynamic light scattering (Delsa™Nano C; Beckman Coulter).
Circular dichroism
Peptides were dissolved in 5 mM Tris-HCl buffer at a final concentration of 0.1 mg/mL in 0.1% (v/v) DMSO. Circular dichroism (CD) spectra were acquired on a Chirascan™ Spectrometer (Applied Photophysics, Leatherhead, United Kingdom). For measurement in the presence of siRNA, peptides were first prepared at a concentration of 0.1 mg/mL as before, and the samples were titrated with additional of siRNA solution. All the spectra were recorded from 260 to 190 nm (far-UV region) using a 0.5 mm path length cell. The data were processed using Chirascan software where a spectrum of the peptide-free solution was subtracted and Savitzky-Gorlay smoothing applied.
siRNA transfection
A549 cells (human alveolar epithelial adenocarcinoma) were obtained from ATCC (Manassas, VA). The cells were cultured with DMEM supplemented with 10% v/v FBS and 1% v/v antibiotic-antimycotic. One day before transfection, the cells were seeded in six-well plates at a density of 1.6 × 105 cells per well. After overnight incubation, the cells were transfected with peptide/siRNA complexes containing 50 pmol of GAPDH siRNA (50 nM). The complexes were prepared in OptiMEM I reduced serum medium at 10:1 and 20:1 ratio (w/w). Lipofectamine 2000 was used as control. The complexes were added to the cells and incubated for 4 h at 37°C before being washed with phosphate buffered saline (PBS). The transfection medium was removed and replaced with serum supplemented cell culture medium. At 72 h post-transfection, the cells were washed and lysed with cell lysis buffer. Western blotting assay was performed to analyze the level of GAPDH protein.
Flow cytometry
Flow cytometry was used to investigate the cellular uptake of peptide/siRNA complexes. A549 cells were seeded in six-well plates at a density of 2.5 × 105 cells per well 1 day before the experiment. The cells were transfected with peptide/siRNA complexes at 20:1 ratio (w/w) containing 150 pmol of fluorescently labeled siRNA in Opti-MEM I reduced serum medium per well. The transfection medium was removed after 4 h of incubation at 37°C, and the cells were washed with PBS once. The cells were trypsinized by 0.25% (w/v) trypsin-EDTA and suspended in culture medium. The extracellular florescence signal was quenched with 0.04% (w/v) trypan blue solution. After 2 min of incubation, the cells were washed with PBS twice. The cells were resuspended in 500 μL of PBS and sieved with a sterile 40 μm cell strainer (BD Biosciences). The fluorescence intensity was analyzed by flow cytometry (BD FACSCantoII Analyzer; BD Biosciences). At least 10,000 single cells were analyzed for each sample.
Cytotoxicity study
A549 cells were seeded in 96-well plates at a density of 3 × 104 cells per well the day before experiment. Peptide/siRNA complexes prepared at 10:1 and 20:1 ratio (w/w) containing 10 pmol of negative control siRNA in OptiMEM I reduced serum medium per well (50 nM) were added to the cells. After 4 h of incubation at 37°C, the transfection medium was replaced by cell culture medium. MTT assay was carried out at 24 h post-transfection. MTT solution (0.8 mg/mL) was added to the cells. After 2 h, the insoluble formazan was dissolved in isopropanol, and the absorbance at 570 nm was measured. Cell viability was expressed as the percentage of the absorbance from cells treated with complexes against the absorbance from the cells in Opti-MEM I reduced serum medium.
Results
siRNA binding
The siRNA binding affinity of the peptides was examined by gel retardation assay (Fig. 1). The gradual disappearance of siRNA band indicated the interaction between the peptide and the nucleic acids. Complete siRNA binding of KL4 occurred at around 20:1 w/w ratio at which the siRNA band was no longer visible. Compared to the parent KL4 peptide, KA1, KA2, and KV4 showed improved siRNA binding affinity as the disappearance of siRNA band occurred at a lower ratio of around 10:1. In contrast, KA3 and KA4 failed to bind with siRNA at 30:1 ratio, which was the highest ratio tested in the study. The release profile of the siRNA from the complexes was further investigated for the peptides that showed successful binding, that is, KL4, KA1, KA2, and KV4 peptides (Fig. 2). The re-appearance of the siRNA band occurred when the heparin to siRNA ratio reached 10:1 (w/w) for both KL4/siRNA and KA2/siRNA complexes, indicating the dissociation of the complexes. KA1/siRNA complexes were easily dissociated as the siRNA band could be observed at a lower heparin to siRNA ratio of 2:1 (w/w). On the other hand, KV4 appeared to have the strongest association with siRNA as the intensity of the siRNA band was not fully recovered at the highest heparin concentration used.

siRNA binding study of KL4 peptide and its analogs by gel retardation assay. Peptide/siRNA complexes were prepared at 2:1, 5:1, 10:1, 15:1, 20:1, 25:1, and 30:1 ratio (w/w). Naked siRNA was used as control. Electrophoresis was carried out at 125 V for 25 min, and the gel was visualized under UV illumination. siRNA, small interfering RNA; UV, ultraviolet.
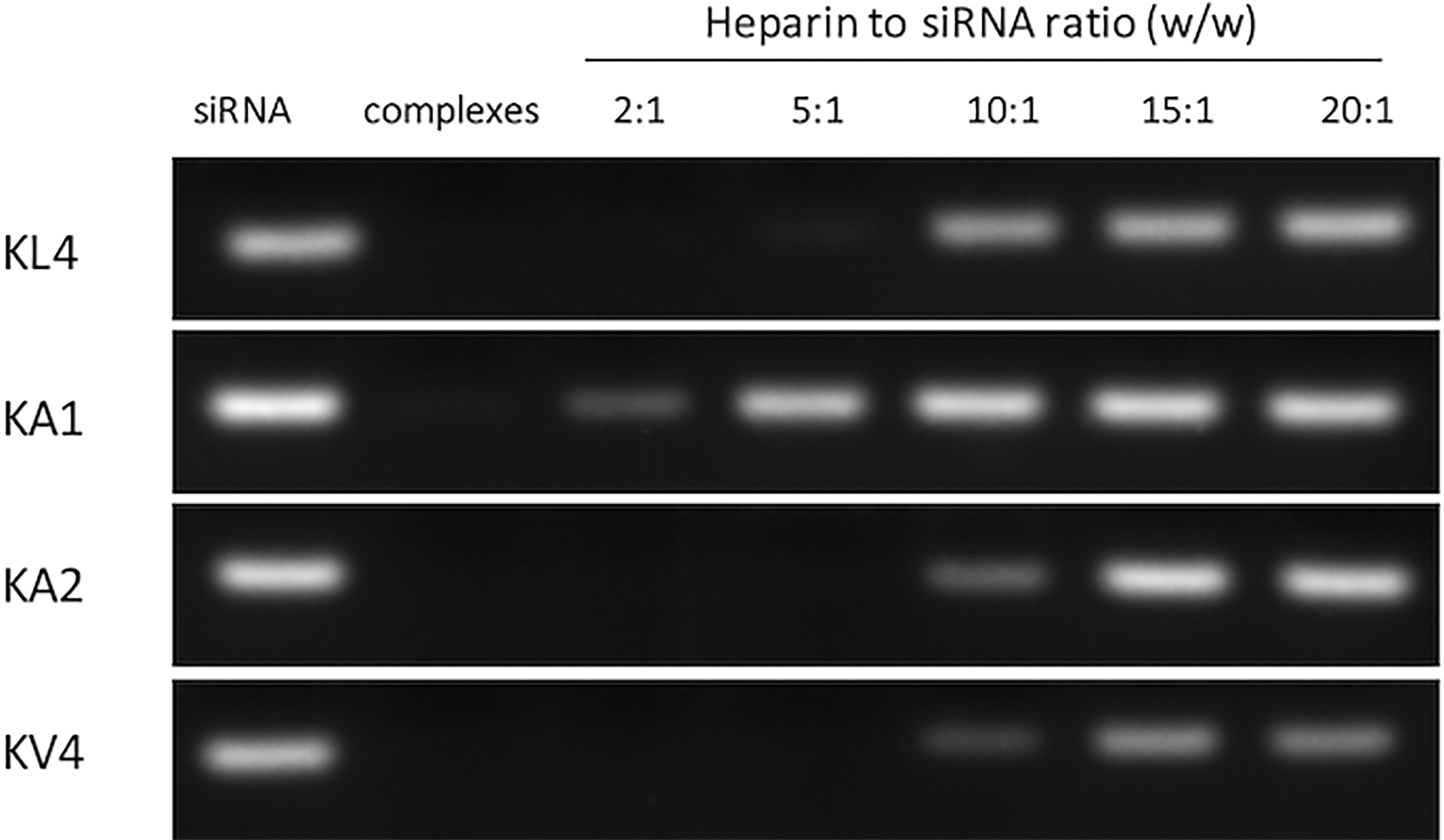
siRNA release study by gel retardation assay. Peptide/siRNA complexes were prepared at ratio 20:1. Heparin was added to the complexes at various heparin to siRNA ratio from 2:1 to 20:1. Naked siRNA was used as control. Electrophoresis was carried out at 125 V for 25 min, and the gel was visualized under UV illumination.
Particle size distribution
The particle size of the peptide/siRNA complexes was measured by dynamic light scattering (Table 2). The diameter of the KL4/siRNA complexes was around 158 nm, which was the smallest among all the tested complexes. KA1/siRNA complexes had the largest particle size with diameter over 4,000 nm, which was significantly larger than other complexes (P < 0.0001), while the KA2 and KV4/siRNA complexes were similar in size with diameter of around 300 nm. The size data could not be obtained for the KA3 and KA4 peptide as they failed to bind with siRNA to form complexes. The polydispersity index (PDI) of the KA2 and KV4/siRNA complexes was similar with a value around 0.25, while the PDI was 0.33 and 0.43 for KL4/siRNA and KA1/siRNA complexes, respectively.
Particle Size of Peptide/Small Interfering RNA Complexes
All the complexes were prepared at ratio 20:1 (w/w). The data are presented as mean ± standard deviation (n = 3). The data were analyzed by one-way analysis of variance followed by Tukey's post-hoc test.
p < 0.0001 between KA1/siRNA complexes and other complexes.
siRNA, small interfering RNA.
Secondary structure of peptides and siRNA
The secondary structures of the peptides were examined at room temperature using CD measured at the far-UV region (Fig. 3). KL4 adopted an α-helical conformation as the typical strong positive band at 190–195 nm and two negative bands at 208–210 and 222 nm were observed in the CD spectrum. KV4 appeared to adopt a β-sheet conformation as demonstrated by a single negative band between 210 and 225 nm, and a positive band between 190 and 200 nm. The observation of α-helical conformation with KL4 and the β-sheet conformation with KV4 in aqueous environment suggests that both peptides are likely to be self-aggregating. The rest of the analogs that contained alanine residues in the sequence lost most of the α-helical conformation by adopting more disordered conformations with some β-turn character, as suggested by the low intensity of both positive and negative bands in the CD spectra. Selected peptides were further examined for their structure in the increasing amount of siRNA (Fig. 4). For all the three peptides investigated, namely KL4, KA2, and KV4, the intensity of the positive and negative bands decreased, which might indicate an increase in disorder and/or a modest contribution from the siRNA to the far-UV CD. All three peptides retained a reference for their starting conformation. At peptide/siRNA ratio of 20:1 w/w, the α-helical feature of KL4 peptide could still be observed although the intensity of the positive band and both negative bands diminished. Similarly, the β-sheet structure of KV4 became less prominent as the amount of siRNA increased. For KA2, the presence of siRNA had the least impact on the peptide structure as the β-turn was already dominant before the addition of siRNA.

Far-UV CD spectra of peptides measured at room temperature. The samples were prepared in 5 mM Tris-HCl buffer. Spectra were recorded from 190 to 260 nm using a 0.5 mm pathlength and were processed using Chirascan software where a spectrum of the peptide-free solution was subtracted and Savitzky-Gorlay smoothing applied. CD, circular dichroism. Color images are available online.
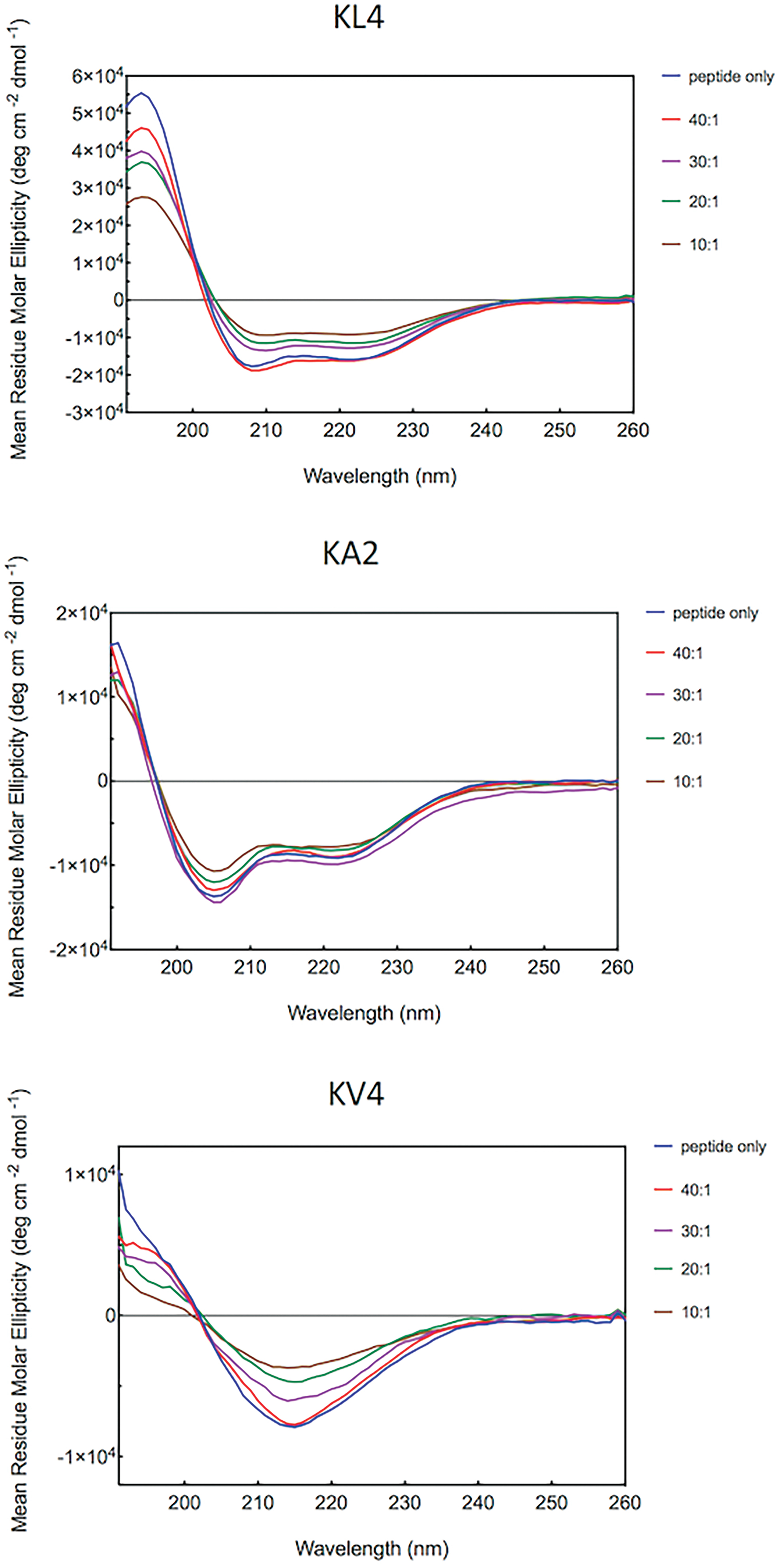
Far-UV CD spectra of peptides titrated with siRNA at room temperature. KL4, KA2, and KV4 peptides were prepared at a concentration of 0.1 mg/mL and was titrated with the addition of small volumes of siRNA solution from ratio 40:1 to 10:1 (w/w). Spectra were recorded from 190 to 260 nm using a 0.5 mm pathlength cell and were processed using Chirascan software where a spectrum of the peptide-free solution was subtracted and Savitzky-Gorlay smoothing applied. Color images are available online.
siRNA transfection, cellular uptake, and cytotoxicity
The siRNA transfection of the peptides was performed on A549 cells using siRNA targeting GAPDH (Fig. 5). Among all the peptides investigated, only KL4 showed successful siRNA transfection with inhibition of GAPDH expression at both 10:1 and 20:1 w/w ratios. All the modified peptides failed to mediate siRNA transfection at the tested ratios. The cellular uptake of peptide/siRNA complexes on A549 cells was also investigated on selected peptides with fluorescently labeled siRNA using flow cytometry (Fig. 6). KL4 peptide had a percentage of cellular uptake of more than 70%. Interestingly, gene silencing effect of some complexes was not reflected by their cellular uptake efficiency. Despite its poor gene silencing effect, KV4 demonstrated a high cellular uptake with percentage of cell uptake of over 80%, which was significantly better than KL4 (P < 0.01). In contrast, KA2/siRNA complexes were not taken up by the cells at all. MTT assay was performed on A549 cells to evaluate the cytotoxicity of the peptide/siRNA complexes prepared at 10:1 and 20:1 w/w ratios (Fig. 7). In general, there was no significant difference among all the groups investigated, and there was no sign of cytotoxicity of the complexes at the investigated ratios.
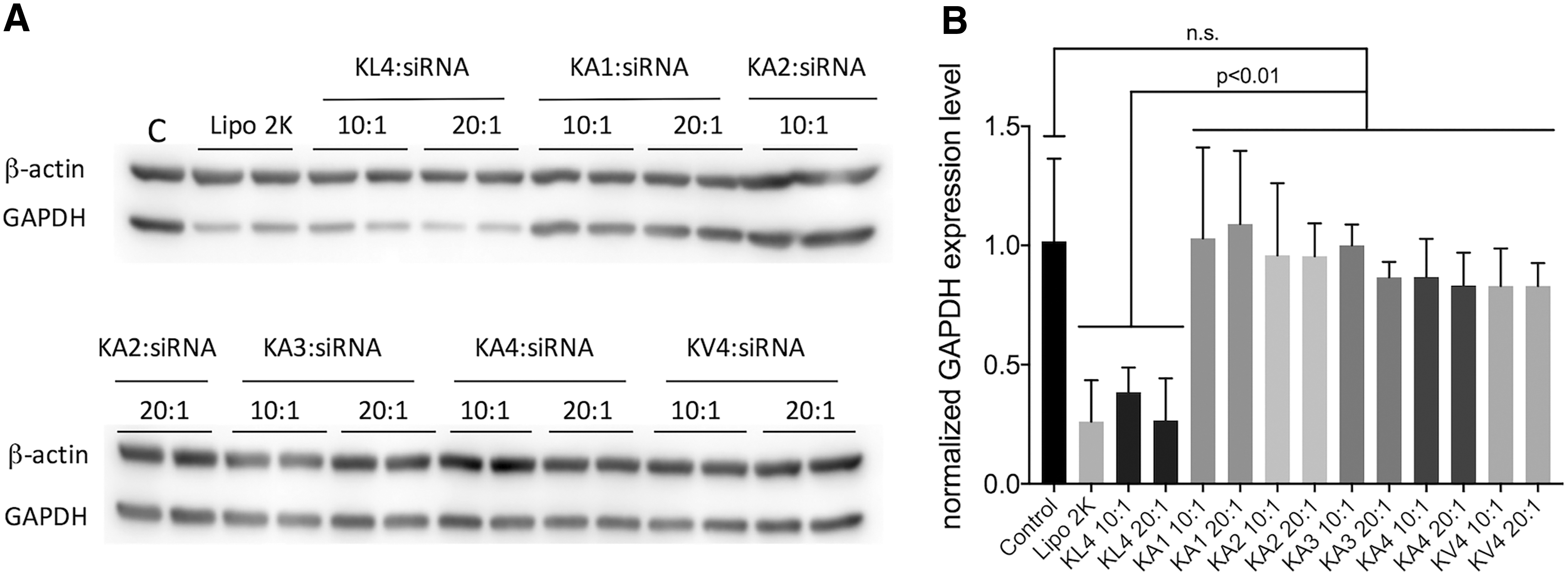
siRNA transfection of peptides on A549 cells. Peptide/siRNA complexes were prepared at 10:1 and 20:1 w/w ratios with 50 pmol of GAPDH siRNA per well in a six-well plate (50 nM siRNA). Lipofectamine 2000 (Lipo 2k)/siRNA complexes at 2:1 v/w ratio was used as positive control.

Cellular uptake study using flow cytometry. A549 cells were treated with peptide/siRNA complexes prepared at 20:1 ratio (w/w) with 150 pmol fluorescently labeled siRNA per well in a six-well plate. Cells were examined at 4 h post-transfection using flow cytometry.
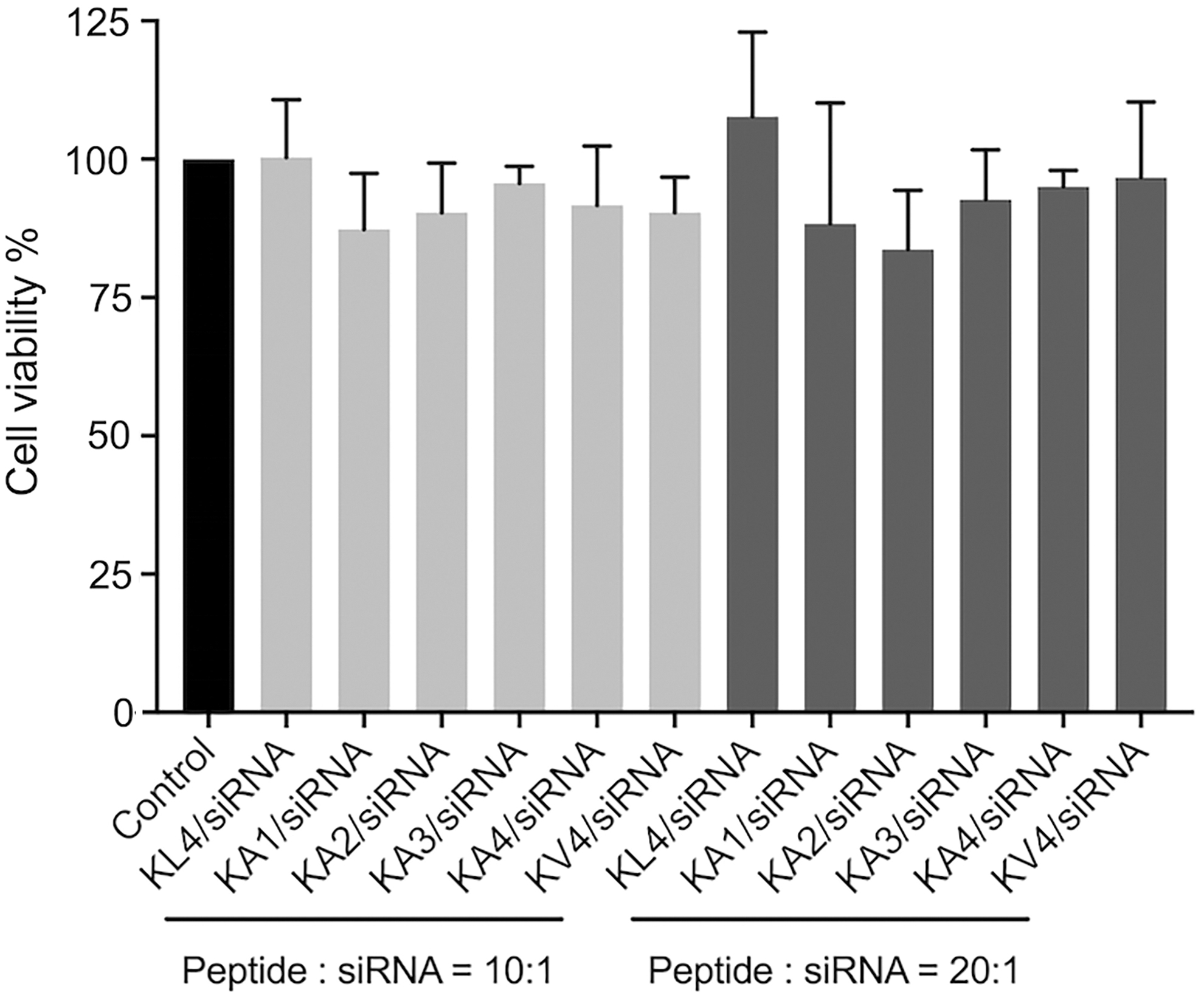
MTT cytotoxicity study of peptide/siRNA complexes on A549 cells at 24 h post-transfection in a 96-well plate. Peptide/siRNA complexes were prepared at 10:1 and 20:1 ratios (w/w) with 10 pmol of siRNA in OptiMEM (control) per well (50 nM of siRNA). Results were presented as absorbance of treated cells relative to the absorbance of control. Values are the mean ± standard deviation of three independent repeats (n = 3). MTT, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide.
Discussion
While the cationic KL4 peptide has been demonstrated to be efficient in transfecting siRNA in vitro [20], its clinical application is hampered by its low aqueous solubility due to the presence of a high number of hydrophobic leucine residues, which contributed to over 75% of the amino acid residues in the sequence. To address this issue, the KL4 peptide was modified by substituting the leucine residues with alanine residues, either partially or completely. This peptide modification strategy is commonly employed in the design of antimicrobial peptides to manipulate the hydrophobicity to optimize their antimicrobial effect by obtaining a balance between antibacterial and hemolytic activities [21–23].
The siRNA binding affinity of the analogs was examined by gel retardation assay. The formation of complexes between peptides and siRNA relies on the electrostatic interaction. Although the overall charge of the analogs was the same as the parent KL4 peptide (the number and the position of the positively charged lysine residue remained the same as KL4), the binding affinity was very different among the different analogs. It was anticipated that peptides with high hydrophobicity have a tendency to self-aggregate, as observed in some antimicrobial peptides [21], thereby reducing their ability to interact with the nucleic acids. When one or two leucine residues were replaced with the less hydrophobic alanine in each repeating segment (KA1 and KA2), the siRNA binding was indeed improved. However, when the hydrophobicity of the peptides was further decreased by replacing three or four leucine residues with alanine in each segment (KA3 and KA4), the siRNA binding became severely hindered. One of the possible explanations was the difference in the accessibility of the positively charged lysine to the negatively charged siRNA, hence affecting the binding affinity. To explore this concept, CD was used to measure and compare the secondary structures of these peptides. The CD data revealed that KL4 peptide adopted an α-helix conformation, whereas all the alanine-containing peptides lost the helical conformation, becoming more disordered and with a preference for β-turn conformations. This observation was consistent with previous studies on antimicrobial peptides in which the alanine analogs exhibited a reduced α-helix content compared to their leucine or isoleucine counterparts [22,24]. In addition, when leucine was completely replaced with the more hydrophobic valine (KV4), the peptide changed to a β-sheet conformation and the siRNA binding was improved as well. The CD data also suggested that the more hydrophobic KL4 and KV4 peptides are self-associating in the aqueous environment. However, there was no clear correlation between the secondary structure of the peptides and their siRNA binding affinity.
While the conformations of the peptides do not give any meaningful clue about their differences in siRNA binding affinity, they provide useful information about the structural requirement to achieve good siRNA transfection efficiency. Disappointingly, none of the KL4 analogs was successful in transfecting siRNA in cells in vitro. Only the parent KL4 peptide, also the sole peptide with high α-helix conformation, showed effective gene silencing effect. In the presence of siRNA, the α-helix conformation of KL4 could still be detected, although to a lesser extent compared to the free peptide. In fact, peptides designed for siRNA delivery typically have an α-helix conformation, such as CADY and LAH4 [25–28]. It is reported that the α-helix conformation is important for the peptide vector to interact with the plasma membrane either during cell entry or for the promotion of endosomal escape, thereby facilitating nucleic acid transfection. Our result here has confirmed the importance of α-helix structure for effective siRNA delivery, although the nature of interaction between KL4 and the plasma membrane remained to be investigated.
Another intriguing observation in this study was that the cellular uptake did not correlate with the gene silencing efficiency. While KV4 analog was the most effective peptide in mediating siRNA uptake into the cells, it failed to induce any gene silencing effect. In fact, it has been suggested that certain CPPs such as MPG, which adopt a β-sheet conformation, are capable of inserting into the plasma membrane [29]. According to the literature, the insertion of β-sheet structured peptides in the phospholipid environment could temporarily alter the organization of plasma membrane, leading to the creation of a transient channel that allows the peptide/siRNA complexes to enter the cells [25]. This could perhaps explain the high cellular uptake of siRNA mediated by the β-sheet KV4 peptide. Whether KV peptide could retain the β-sheet structure in the lipid environment remained to be investigated.
However, the high cellular uptake did not translate to efficient gene silencing, which could be due to the unsuccessful release of siRNA from the complexes after cell entry. A relatively strong interaction between the KV4 analog and siRNA may hinder the dissociation of the complexes, as demonstrated in the gel retardation assay, hence siRNA was not readily available to induce RNAi in the cytoplasm. It is therefore crucial to strike a good balance between siRNA binding and release to achieve a good transfection efficiency. Another possibility was that the highly hydrophobic residues in KV4 may prevent the peptide from translocating into the cytoplasm, with the peptide being trapped in the membrane due to its strong interaction with the phospholipids [30]. For KA2 analog, it was not surprising to see a poor gene silencing effect as the cellular uptake was extremely low anyway. Although KA2/siRNA and KV4/siRNA complexes had similar particle size of around 300 nm, their performance in terms of cellular uptake was very different, suggesting that uptake efficiency was related to the peptide conformation, rather than the size of the complexes.
Conformational versatility of CPPs has been reported to be crucial for their cellular uptake efficiency [31]. This refers to the ability of the peptide to change its structure in response to the environment such as the cell membrane. Therefore, it is of great interest to examine the structure of the peptides not only in their free form but also in the presence of membrane-mimicking environment. CPPs such as MPG and CADY (which are mentioned earlier) demonstrate the conformational versatility as they exhibit mainly disordered conformation in solution, but adopt β-sheet and α-helix conformation, respectively, in the presence of phospholipid vesicles [29,32]. Previous study on KL4 showed that in the presence of lipids, the peptide can adopt a helical structure, but the backbone torsion angles were different from the common values for α-helix [33,34]. Since the secondary structure of CPP is dynamic, further investigation on the structural conformation of other KL4 analogs in a lipid environment would provide us with more information about their interaction with the cell membrane, and how that might affect their delivery efficiency.
Unfortunately, despite the attempt to increase the peptide solubility by replacing leucine with the less hydrophobic alanine residues, none of the modified peptides was able to dissolve in water at a concentration of 1 mg/mL, hence DMSO (1%) was still required to dissolve the peptide in this study. Since the average hydrophobicity calculated by means of the Eisenberg Consensus scale was positive, indicating all of these of peptides were still hydrophobic in nature. In the future development, the design of sequence is aimed to produce helical structures, with the amino acids arranged in such a way that the positively charged lysine is aligned on the hydrophilic face, whereas the hydrophobic leucine or alanine residues are aligned on the hydrophilic face, as the segregation of the hydrophilic and hydrophobic surfaces is reported to be crucial for effective delivery efficiency [35]. An alternative strategy has been sought to address the solubility issue of KL4 peptide. In fact, our recent study has shown very encouraging results that PEGylation is a promising strategy to develop KL4 peptide for messenger RNA (mRNA) delivery [36]. Not only the aqueous solubility was significantly enhanced but also the mRNA transfection efficiency of PEGylated KL4 was found to be superior to the parent KL4 peptide. Future investigation on the PEGylated peptide structure and its potential for delivering siRNA, as well as other oligonucleotide therapeutics is warranted.
Conclusions
Although the modification of KL4 peptide by substituting the leucine residues with the less hydrophobic alanine failed to achieve satisfactory siRNA transfection, this work asserts the importance of the hydrophobicity and α-helix conformation of surfactant peptides in mediating effective siRNA delivery. With the understanding of the structural requirement, alternative modification strategy can be explored to improve KL4 as a siRNA delivery vector.
Footnotes
Acknowledgments
The authors would like to thank Dr. Alex Drake and Dr. Tam Bui, King's College London for their kind assistance in performing and analyzing the CD measurement.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The work is supported by Seed Fund for Basic Research, The University of Hong Kong (201711159172). Simon Wong Pharm Manufacturing Industrial Scholarship supported Y.Q. for traveling to King's College London for performing the CD experiments in this study.