
Abstract
Antisense-mediated exon skipping constitutes a promising new modality for treatment of Duchenne Muscular Dystrophy (DMD), which is caused by gene mutations that typically introduce a translation stop codon in the dystrophin gene, thereby abolishing production of functional dystrophin protein. The exon removal can restore translation to produce a shortened, but still partially functional dystrophin protein. Peptide nucleic acid (PNA) as a potential antisense drug has previously been shown to restore the expression of functional dystrophin by splice modulation in the mdx mouse model of DMD. In this study, we compare systemic administration of a 20-mer splice switching antisense PNA oligomer through intravenous (i.v.) and subcutaneous (s.c.) routes in the mdx mice. Furthermore, the effect of in situ forming depot technology (BEPO®) and PNA-oligonucleotide formulation was studied. In vivo fluorescence imaging analysis showed fast renal/bladder excretion of the PNA (t½ ∼ 20 min) for i.v. administration, while s.c. administration showed a two to three times slower excretion. The release from the BEPO depot exhibited biphasic kinetics with a slow release (t½ ∼ 10 days) of 50% of the dose. In all cases, some accumulation in kidneys and liver could be detected. Formulation of PNA as a duplex hybridization complex with a complementary phosphorothioate oligonucleotide increased the solubility of the PNA. However, none of these alternative administration methods resulted in significantly improved antisense activity. Therefore, either more sophisticated formulations such as designed nanoparticles or conjugation to delivery ligands must be utilized to improve both pharmacokinetics as well as tissue targeting and availability. On the other hand, the results show that s.c. and BEPO depot administration of PNA are feasible and allow easier, higher, and less frequent dosing, as well as more controlled release, which can be exploited both for animal model studies as well as eventually in the clinic in terms of dosing optimization.
Introduction
Peptide nucleic acid (PNA) is widely used in chemistry and molecular and cell biology applications [1,2], but only limited efforts have been dedicated to in vivo drug discovery studies, although some of these do indicate an interesting potential [3,4]. More than 15 years ago, it was demonstrated that PNA could be used to modulate pre-mRNA splicing in a transgenic mouse model (EGFP-654) using a high dose of Lys4-PNA (4 × 50 mg/kg) [5]. Later, unmodified PNA (and various PNA conjugates) has been reported to restore the expression of functional dystrophin by splice correction by intramuscular (i.m.) administration in mice [6,7]. Furthermore, nanoparticle-formulated PNA has been demonstrated to exhibit gene correction activity and anti-MiR activity in mouse models [3,4,8,9].
Systemic administration [intraperitoneal (i. p.) or intravenous (i.v.)] of unmodified PNA results in fast clearance from the circulation through the kidneys, typically with half-lives <1 h [10–13]. Therefore, to increase PNA in vivo efficacy, the pharmacokinetic profile must be improved. Several studies have demonstrated in vivo activity of splice-switching antisense PNAs in the mdx mouse model of muscular dystrophy, and this model was therefore chosen in this study to investigate in vivo administration routes of PNA. Duchenne muscular dystrophy (DMD) is an X-linked genetic disorder and the most common form of muscular dystrophy. It is caused by gene mutations in the dystrophin gene that leads to severe deficiency of the functional protein. DMD is often fatal before the end of the third decade of life, typically due to respiratory or cardiac failure. In general, only palliative treatments such as glucocorticoids are available, although two new drugs were recently approved, one in Europe (Ataluren) and one in the United States (Eteplirsen) [14,15]. Eteplirsen is an antisense phosphoramidate morpholino oligomer (PMO) targeting exon 51.
The mdx mouse model for DMD has a point mutation in exon 23 of the dystrophin gene that generates a translation stop codon. Antisense oligonucleotide (AON) mediated exon skipping, using AONs targeting the splice junction of exon 23, which induce spliceosomal skipping of this exon to restoring downstream gene translation resulting in a shortened, but functional dystrophin protein. Clinical use of antisense therapy for treatment of DMD requires systemic administration of the drug because all muscles, also including heart and diaphragm muscles, are affected by the disease and must be reached. Some of the most effective PNA conjugates with good activity upon i.m. administration in mdx mice showed far less efficiency when administered systemically [16].
More recently, Gao et al. compared systemic delivery of an unmodified PNA and the corresponding PMO in the mdx mouse model using an identical high dosing regimen (3 × 50 mg/kg intravenously at weekly intervals), and showed comparative systemic activity in peripheral muscles: quadriceps (Quad), gastrocnemius (Gas), triceps, and diaphragm [17]. In the same study, they increased the dosing to 5 × 100 mg/kg at weekly intervals, and achieved high levels of exon skipping in Quad and Gas and up to 40% of normal dystrophin levels assessed by western blotting.
In this study, we used the mdx mice as a model to investigate different administration routes and delivery approaches of PNA, including a novel drug delivery system, BEPO®, which is a long-acting, injectable in situ depot forming technology based on diblock and triblock poly(ethylene glycol)-polyesters solubilized in a biocompatible solvent. The depot formation is then mediated by a solvent exchange mechanism [18], and the delivery profile of the formulated molecule can be modulated by varying polymer composition and polymer content. This technology has the potential to design sustained release formulations of small molecules with release kinetics varying from weeks to months. Furthermore, it can also be used to formulate peptides and therapeutic proteins without affecting protein functionality and activity [19].
Materials and Methods
PNA synthesis
PNA synthesis was carried out by tBoc solid-phase methodology as described previously [20]. The PNAs used in this study share the same base sequence, a 20-mer targeting the junction sequences of exon 23 and intron 23 of the murine DMD gene (PNA3696: H-GGCCAAACCTCGGCTTACTC-NH2), which has previously been shown to induce exon 23 skipping [6,21]. Alexa Fluor 680 (AF680) or Cy5.5 was coupled in solution to a cystein added to the N-terminal of the PNA during solid-phase synthesis through the fluorophore maleimide derivatives. The PNA was purified by high performance liquid chromatography (HPLC) and characterized by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (Supplementary Fig. S1), lyophilized, and stored at 4°C until use.
Preparation of PNA administration solutions
PNA stock solutions were prepared in Milli-Q water. AF680- or Cy5.5-labeled PNAs were dissolved in a droplet of dimethyl sulfoxide (DMSO) and then water. The PNA concentration (in mg/mL) was calculated based on molar concentration measured spectrophotometrically and using the molecular weight of the PNA. This was done due to variable trifluoroacetic acid (TFA)-salt content in the PNA preparations [by protonation of the nucleobases (predominantly cytosine and adenine)]. Therefore, a PNA solution in pure H2O is very acidic (typically pH <1–2, depending on the PNA concentration). Solutions for animal administration were prepared in isotonic glucose and adjusted to approximately pH4 with 1 M NaOH. When adjusting the pH of the injection solution, local precipitation was observed upon the addition of a drop of 1 M NaOH. At pH higher than 4, sustained precipitation in the injection solution occurred at a PNA concentration of ∼1 mM corresponding to an administration solution used for a dose of 50 mg/kg. In addition, injection of this administration solution into serum caused significant visible precipitation (Supplementary Fig. S2). The injection volumes for i.m. and i.v. administrations were 10 and 200 μL, respectively. Hybridization of PNA-DNA and PNA-phosphorothioate oligonucleotide heteroduplexes was performed in citrate buffer (pH7) at 95°C to avoid precipitation of the PNA before the addition of the DNA and phosphorothioate oligonucleotides (Integrated DNA Technologies, Coralville).
BEPO-PNA3696 formulations
Diblock (DB) mPEG-PDLLA and triblock (TB) PDLLA-PEG-PDLLA copolymers were synthesized by MedinCell. USP grade DMSO was purchased from Gaylord Chemical (Los Angeles, CA). Table 1 describes the copolymers that were used during the formulation screening in terms of molecular weights of the PEG or mPEG and PDLLA moieties composing the copolymer molecule.
Description of the Copolymers Present in BEPO-PNA Formulations
PDLLA, poly-
Setup of HPLC method for PNA3696 detection and quantification
PNA3696 was analyzed by RP-HPLC (PDA detector; Waters Alliance System) using a Widepore Aeris 3.6 μm; 150 × 4.6 mm; XB-C18 column (Phenomenex, Torrance). A gradient elution method was applied with mobile phases A: 0.1% (v/v) TFA in acetonitrile (ACN) and B: 0.1% (v/v) TFA in water. A 5%–28% linear gradient of eluent A in 5 min was used and delivered at a 1 mL/min flow rate. Column temperature was set at 60°C and ultraviolet absorbance was measured at 260 nm. The retention time of PNA molecule was 4.62 min. A minor impurity accounting for 1.2% relative area and presenting a relative retention time (RRT) of 0.92 could be observed. Method linearity was demonstrated over a 2- to 100-μg/mL concentration range. Quality control samples within the concentration range were used to validate precision and accuracy of the method.
Preparation of BEPO-PNA3696 formulations
Vehicles made of TB and DB copolymers were prepared at least 1 day ahead before addition of the PNA. Copolymers were weighed and dissolved in DMSO to obtain the targeted polymer content in the vehicle, generally comprised between 35% and 40% (w/w). The sample was left to mix overnight on roller mixer at room temperature until complete polymer solubilization. Once homogeneous solution was obtained, PNA3696 lyophilized powder presenting a PNA content of roughly 50% (w/w) was weighed and added into the vehicle to reach, in general, a PNA loading of 0.25% to 0.5% (w/w) within the final formulation. For some in vivo studies, PNA loading was increased up to 2% (w/w). PNA was soluble in the final formulation, in agreement with the fact that the maximal solubility of PNA in DMSO had been measured at 56 mg/mL. AF680 was not compatible with the BEPO formulation; therefore, Cy5.5 was chosen instead. Formulations for in vivo studies were sterile filtered before reconstituting the formulation under laminar flow hood. PNA concentration in the formulation was determined by weighing accurately around 50 mg of formulation in a 10-mL volumetric flask. After addition of 1 mL of ACN and vigorous shaking to solubilize the polymer, water with 0.1% (v/v) TFA was added up to the mark. An aliquot was taken, centrifuged 5 min at 13,000 rpm, and analyzed by HPLC for PNA quantification and ultimate determination of PNA loading in the formulation.
In vitro release studies
In vitro release (IVR) studies of BEPO-PNA3696 formulations were performed in Krebs Ringer Tris (KRT) buffer at pH7.4. Depots were formed by direct injection of the formulation into the release medium and placing at 37°C under gentle orbital shaking. One hundred milligrams of formulation corresponding to 250–500 μg of PNA was injected into 10 mL of buffer, ensuring sink conditions as PNA solubility in KRT had been measured at 440 μg/mL. At regular time points, the entire release medium was replaced by fresh buffer and PNA in the sampled buffer was quantified by HPLC. Stability of a PNA solution at 10 μg/mL in KRT was at least 3 weeks at 37°C. PNA loading, polymer content, and composition were the main parameters that were investigated for the IVR screening to modulate release kinetics. At the end of the IVR study, the polymer depot was solubilized in 1.5 mL of ACN. After polymer solubilization, 13.5 mL of H2O + 0.1% (v/v) TFA was added and the sample was centrifuged 5 min at 13,000 rpm. The supernatant was then analyzed by HPLC to determine the PNA amount remaining in the depot.
In vivo experiments
Animals
All animal experiments were performed in accordance with the regulations stated in the European Convention for the Protection of Vertebrate Animals used for Experimentation, with permission from the Danish Animal Experiments Inspectorate. The mice were housed in individually ventilated cages in a temperature-controlled environment with a 12-h light/12-h dark cycle and provided with a rodent chow diet and water ad libitum.
PNA treatment studies in mdx mice
Eight- to 9-week-old mdx male mice (bred at the animal facility at University of Copenhagen, Denmark) were injected subcutaneous (s.c.) or i.v. with PNA3696 with semiweekly dosing (n = 4), or injected s.c. with either BEPO-PNA3696 or vehicle formulation, with either 2 (65–70 μL) or 3 depots (∼90 μL) corresponding to ∼100 or 200 mg/kg PNA3696 per mouse, respectively (n = 5). The mice for i.m. injections were anesthetized by inhalation of isoflurane (Baxter, Deerfield, IL) and 10 μL of BEPO-PNA formulation (n = 6) or PNA heteroduplex solution (10 μg, n = 4) were injected into tibialis anterior (TA) muscles along the fiber orientation using an insulin syringe. All mice were euthanized by cervical dislocation at desired time points, after which the TA, Gas, Quad, soleus (Sol) and extensor digitorum longus (Edl) muscles were collected and snap-frozen in liquid nitrogen-cooled isopentane, and stored at −80°C.
In vivo imaging
Six- to 10-week-old female NMRI-nu (Rj:NMRI-Foxn1nu/nu) mice purchased from Janvier Labs (Le Genest-Saint-Isle, France) were used for the in vivo imaging studies. An epi-illumination prescan of the mouse was performed in an IVIS spectrumCT in vivo imaging system (PerkinElmer, Waltham, MA) using an ex. 675 nm/em. 720 nm filter set. PNA was then administrated either i.v. or s.c. (200 μL) and the mice were scanned at different time points (n = 2 or 3 per time point). To evaluate the BEPO-PNA3696 release kinetics, a formulation with 1% PNA loading [Cy5.5-labeled PNA3696 and PNA3696 (1:100)] or a vehicle formulation was injected s.c. at either ∼45 or ∼90 μL corresponding to a dose of 20 or 40 mg/kg (n = 3–4). All mice were euthanized by cervical dislocation after the last scan, and subsequent ex vivo organ scans were performed. Living Image® software was used for data analysis (PerkinElmer). To quantify the fluorescence signals from the body and kidneys, boundaries were drawn around the region of interest (ROI) to obtain the radiant intensity contained within. Using the radiant intensity corresponding to the first three time points, liner regression analysis was performed with GraphPad Prism (GraphPad Software, San Diego, CA) to determine the initial ROI values (Y0). All ROI values were then converted into percentages based on the estimated initial signal and plotted into GraphPad Prism for the determination of the compound elimination “half-life.” For the BEPO-Cy5.5-PNA-PNA3696 release study, “half-lives” were calculated using the best linear fit from t½ = ln(2)/k.
Dystrophin staining
Frozen, serial sections of 10 μm were cut and mounted on Superfrost Ultra Plus™ (Thermo Scientific, Waltham, MA) microscope slides. The intervening sections were collected for reverse transcription - polymerase chain reaction (RT-PCR). Unfixed sections were blocked in buffer (3% fetal calf serum in phosphate-buffered saline) before staining. Sections were incubated overnight at 4°C with anti-dystrophin (ab15277 at 1:500; Abcam, Cambridge, United Kingdom). Sections were then incubated for 1 h at room temperature with AlexaFluor 594 anti-rabbit (at 1:500; Life Technologies, Carlsbad, CA). Images were taken with a 10 × plan apo objective on a Nikon Eclipse 50i with a Nikon 5-megapixel camera using NIS Elements software (Tokyo, Japan).
Hematoxylin and eosin staining
For histology analysis, three mice (one from each group; 45- and 90-μL Cy5.5-PNA3696 depots and 90-μL vehicle depot) were sacrificed after the 24-h scan. Skin, depot, and muscle underneath were removed and fixed in 4% paraformaldehyde in sodium phosphate buffer, before embedding in paraffin, and stained with hematoxylin and eosin. The samples were examined by a pathologist (blinded) for morphological changes.
RT-PCR
Total RNA was extracted from TA, Gas, and Quad muscles using the TRIzol reagent (Life Technologies) according to the protocol supplied by the manufacturer. cDNA was generated using 1,000 ng of total RNA with the Maxima Reverse Transcriptase (Life Technologies) and the Ex26 RT primer (5′-AGTCTGTAATTCATCTGGAG-3′). Subsequently, 2 μL of cDNA was used for each 25-μL PCR reaction with the DreamTaq DNA polymerase (Life Technologies). The forward and reverse primer sequences were m22f: 5′-ATCCAGCAGTCAGAAAGCAAA-3′ and m24r: 5′-CAGCCATCCATTTCTGTAAGG-3′. The cycling conditions were 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s for 32 cycles. For the detection of low percentages of exon-skipping, the number of cycles was increased to 38. The PCR products were separated in a 2% agarose gel with 1 × TBE buffer and stained with ethidium bromide.
Protein extraction and western blotting
Fifteen sections (30 μm) from Quad muscles were homogenized in ice-cold lysis buffer with protease and phosphatase inhibitors (10 mM Tris, pH7.4, 0.1% Triton-X 100, 0.5% sodium deoxycholate, 0.07-U/mL aprotinin, 20-μM leupeptin, 20-μM pepstatin, 1-mM phenylmethanesulfonyl fluoride, 1-mM EDTA, 1-mM EGTA, and 1-mM DTT) using a bead mill at 4°C. Supernatants were collected and separated on 4%–15% TGX polyacrylamide gels (Bio-Rad, Hercules, CA) at 200 V for 30 min. Proteins were transferred to polyvinylidene fluoride membranes, which were blocked in Baileys Irish Cream (Dublin, Ireland) for 30 min and incubated overnight with dystrophin antibody (ab15277; Abcam). Secondary antibody coupled with horseradish peroxidase diluted 1:10,000 was used to detect primary antibodies (Agilent, Glostrup, Denmark). Immunoreactive bands were detected using Clarity (Bio-Rad) and visualized on a Chemi-Doc MP digital darkroom (Bio-Rad).
Results
I.v. administration
To evaluate the systemic activity of PNA3696 (20-mer unmodified PNA), 8- to 9-week-old male mdx mice received five times 50 mg/kg PNA3696 or vehicle (isotonic glucose) dosed semiweekly by i.v. injection (n = 4). Two weeks after the last injection, muscles where excised and immunohistochemistry, RT-PCR, and western blotting analyses were performed. Surprisingly, and contrary to published data with the same total dose [17], but different dose regimen [five weekly injection (5 weeks) vs. five semiweekly intervals in this study (2½ week)], only low levels of weakly stained dystrophin-positive fibers where observed in gastrocnemius and to a lesser extent in quadriceps muscles (Fig. 1a) of the treated mdx mice. Exon skipping was detectable at the RNA level, but also at very low levels (Fig. 1b). Dystrophin protein was below detection level when analyzed by western blotting (Fig. 1c). To obtain information about the pharmacokinetic profile of unmodified PNA, AlexaFluor 680 (AF680)-labeled PNA3696 (PNA4948) was injected i.v. into female NMRI-nu mice and scanned at different time points in the IVIS Spectrum CT scanner. In a separate experiment, AF680-labeled PNA was mixed with PNA3696 (10, 25, or 50 mg/kg) to emulate the distribution at treatment doses. From dorsal images (Fig. 1d) a gradual decrease in whole-body fluorescence was observed over time for all treatment doses with a very weak residual signal at 24 h. PNA was detected in the kidneys and bladder already after 15 min (Supplementary Fig. S3a), and the elimination half-life based on the IVIS scans for AF680-PNA was ∼20 min (n = 3). Body half-lives at 10, 25, and 50 mg/kg treatment doses were ∼24, 28, and 33 min, respectively (n = 2–3) (Fig. 1e). The ratio between the fluorescence signal from the kidneys and the whole body increased over time (Fig. 1f), demonstrating relatively slower elimination (relative accumulation) of PNAs from the kidneys over time, and this relative accumulation increased with PNA dose. At the highest dose, liver accumulation was clearly visible already after 15 min. Ex vivo organ scans supported dose-dependent accumulation in the kidneys and the liver seen by live imaging (Fig. 1g), with the highest fluorescence signal in the kidneys. To validate the imaging data, we also tested an analogous Cy5.5 PNA conjugate. The distribution and clearance pattern was very similar to that obtained with AF680-PNA, but somewhat higher liver accumulation for Cy5.5 was observed (as also seen by others [22]). Cy5.5 is more lipophilic than AF680, and therefore would be expected to have a larger influence on pharmacokinetics by nonspecific protein binding [23] (Supplementary Fig. S3b, c).
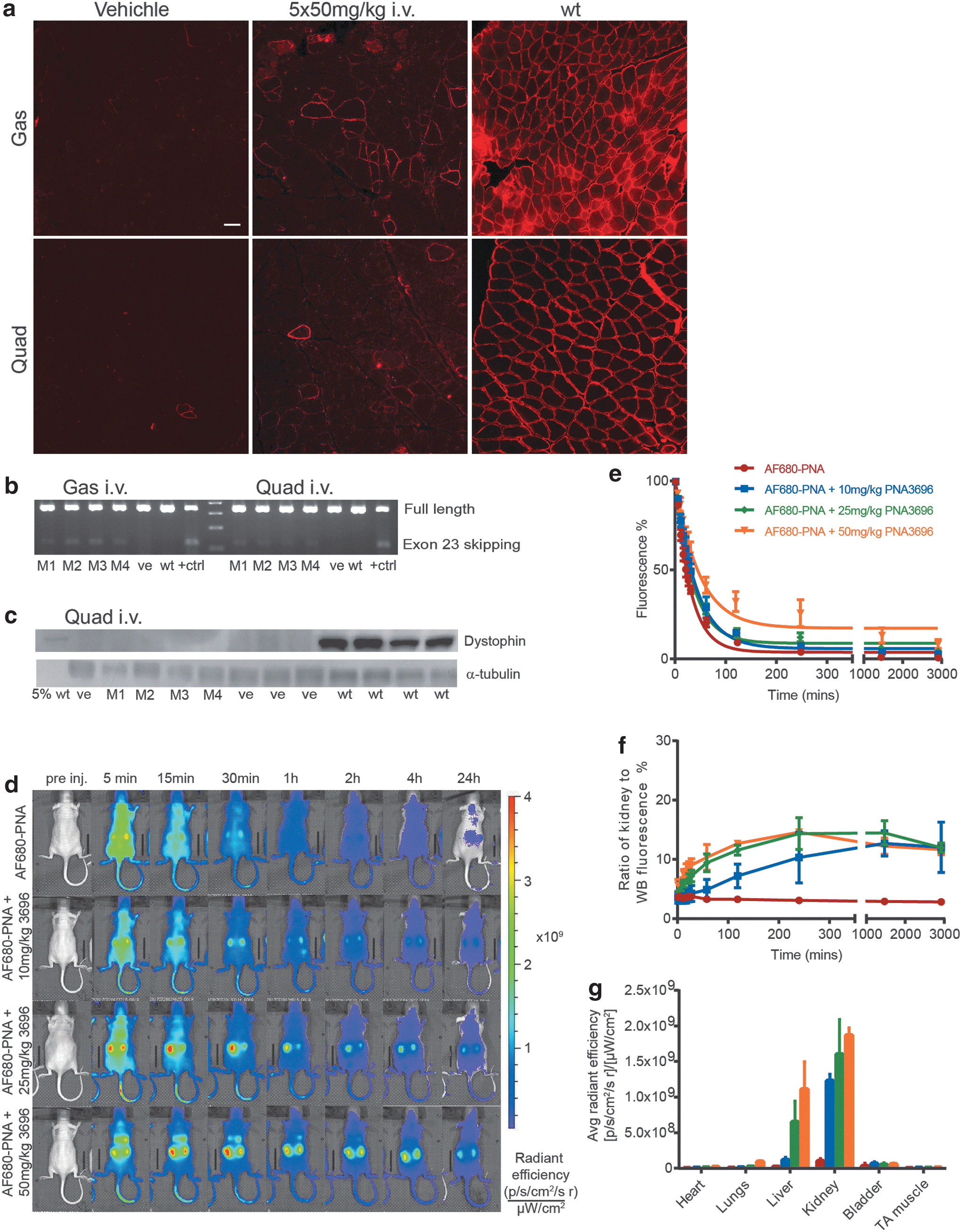
Intravenous administration of PNA in mice.
S.c. administration
To obtain a prolonged body exposure to the PNA (as well as possibility of higher dosing) and thus a more favorable pharmacokinetic profile, the s.c. administration route was evaluated. The images presented in Fig. 2a were obtained using AF680-labeled PNA alone and an s.c. treatment dose of 50 mg/kg PNA (spiked with AF680-PNA). They showed a depot-like structure with slow decrease in fluorescence signal over the first 4 h (almost linear, Fig. 2b). AF680-PNA was detectable in the bladder after 10 min and AF680-PNA in systemic circulation (measured by fluorescence intensity in the tail) peaks after 60 min (Fig. 2c), regardless of treatment dose, and intact AF680-PNA was detected in the urine (at 60-min time point) (Supplementary Fig. S4). Ex vivo organ scans demonstrated accumulation mainly in the kidneys (Supplementary Fig. S5), analogous to i.v. administration. To study the efficacy using this administration route in mdx mice, an s.c. dosing study was performed in 8- to 9-week-old mdx male mice with 5 × 50 mg/kg with semiweekly dosing for two and a half weeks (n = 4). However, only low levels of dystrophin-positive fibers (similar to 5 × 50 mg/kg i.v. study) were observed in gastrocnemius, quadriceps, and TA muscles 2 weeks after the last injection (Fig. 2d), and comparably low levels of exon skipping was confirmed by RT-PCR (Fig. 2e). Western blot analysis of dystrophin protein in the treated muscles was below detection limit (Fig. 2f), although very faint bands were observed when the exposure time was increased. An increased dose of 5 × 100 mg/kg s.c. with semiweekly dosing did not result in higher efficacy (data not shown).

Subcutaneous administration of PNA in mice.
BEPO-PNA formulations and IVR
In an effort to design sustained release formulations, the injectable BEPO drug delivery technology, which is an in situ forming depot method based on the use of PEG-PLA biodegradable copolymers, was used for formulating PNA3696. BEPO-PNA3696 formulations were made with different polymer compositions. A PNA loading of 0.25% (w/w) and polymer content of 40% (w/w) was initially fixed, and different TB and DB molecules (Table 1) were used in a 1:4 TB to DB ratio. In vitro PNA release from the polymer depot was followed at 37°C and varying release kinetics for the different formulations within a 5- to 30-day release duration were obtained (Fig. 3a). Formulation F10 was chosen, and the polymer content was gradually decreased to improve injectability and to allow a formulation injection through a 25G needle or smaller. However, polymer content reduction from 40% to 30% (w/w) resulted in an increase of the formulation burst at day 1 from 7.2% to 32.1% (Fig. 3b). Nevertheless, F16 [35% (w/w) polymer content] was selected as a good compromise and it was further demonstrated that the increase of PNA loading from 0.25% to 0.5% (w/w) in the F16 context did not change the IVR profile of the formulation (Supplementary Fig. S6a). Therefore, formulation F16 was further selected for in vivo evaluation. The formulation could be made several days before animal dosing as an early-stage stability study had shown that PNA was stable in the F16 formulation stored at least 3 weeks at room temperature (Supplementary Fig. S6b).
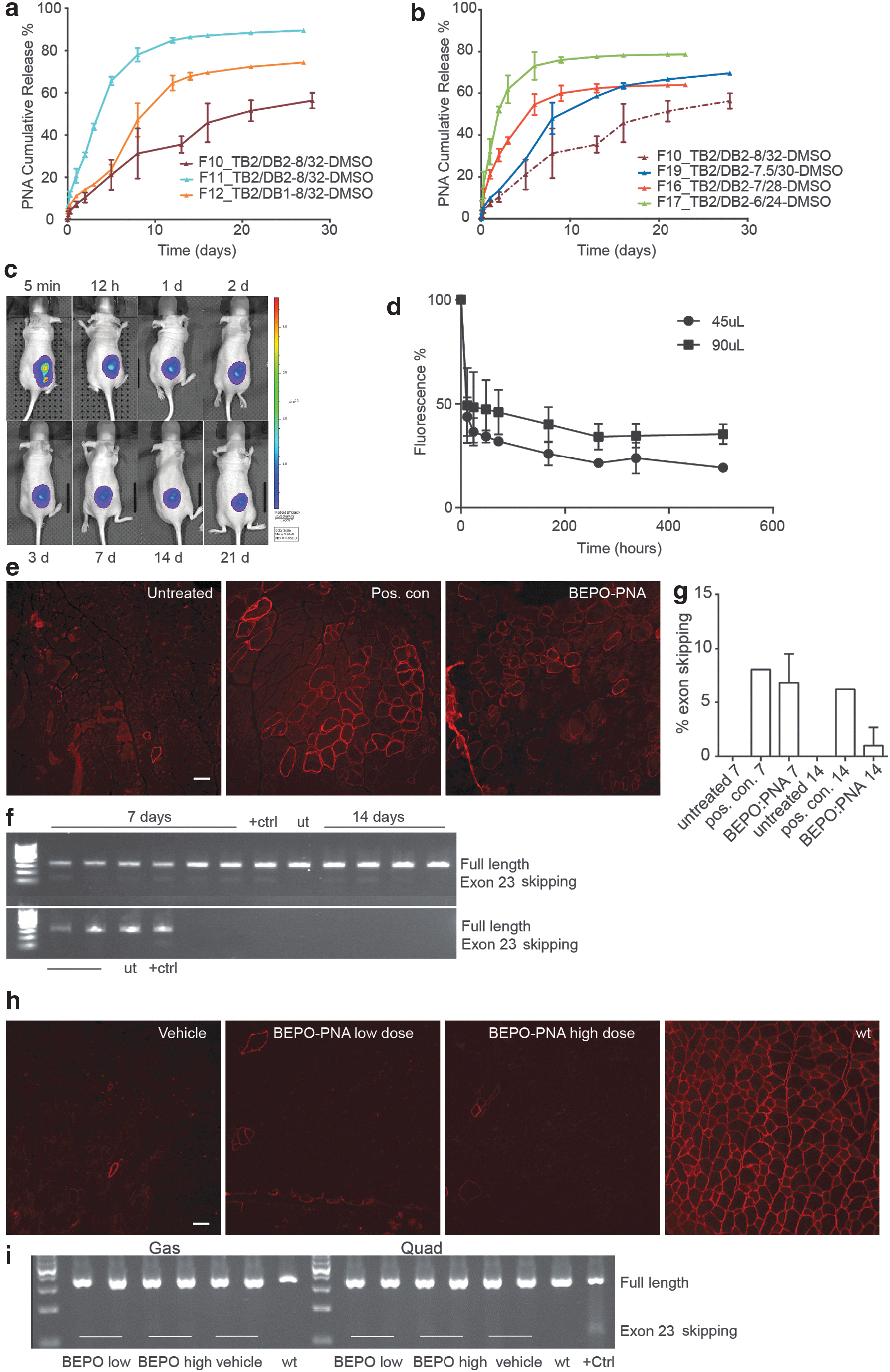
In vitro and in vivo release of PNA from BEPO-PNA formulations. In vitro release profiles of BEPO-PNA formulations.
In vivo administration of BEPO-PNA formulation
A BEPO-PNA formulation with Cy5.5-labeled PNA3696 (PNA4437) and PNA3696 in a 1:100 ratio [1% (w/w) final PNA loading] or polymer vehicle formulation was injected s.c. as either a 45-μL or a 90-μL depot (corresponding to ∼20 mg or 40 mg/kg) on the flank of female nu-NMRI mice and were scanned at different time points (Fig. 3c). (Cy5.5 was used in this case instead of AF680, because of its more hydrophobic nature, which should provide a slower release from the polymer depot.) The Cy5.5-PNA release from the BEPO depot was biphasic (Fig. 3d). The first phase was fast with more than half of the Cy5.5-PNA released within the first 12 h, whereas the second phase was much slower (t½ estimated to ca. 30 days) for both depot sizes. The initial rapid release may occur while the depot is forming in situ through a solvent exchange mechanism. Analysis of the tail showed that it was possible to detect Cy5.5 PNA in systemic circulation up to 21 days postinjection, although at very low levels (Supplementary Fig. S7a). Three mice were sacrificed after 24 h to look for morphological changes in the skin and muscle above and underneath the depots. Neither redness nor any other sign of inflammation was observed macroscopically. Preliminary histological analysis of BEPO-PNA3696 and BEPO-vehicle skin samples indicated only minor acute inflammation with a brim of neutrophil cells around the depots (Supplementary Fig. S7b, c), as would be expected from foreign object implants. No changes in the muscle samples and control samples from untreated mice were observed.
The activity of the BEPO-formulated PNA3696 was initially tested by intramuscular administration. Ten microliters of BEPO-PNA formulation [0.5% (w/w) loading, corresponding to ∼50 μg PNA/TA] was injected into TA muscle in 12-week-old mdx mice and muscles with depots were harvested after 7 and 14 days (n = 6). Dystrophin-positive fibers were observed in all BEPO-PNA3696-treated muscles around the depots (Fig. 3e) similar to the positive control injected with 10 μg PNA3696, supporting full functionality of the PNA after the formulation and release processes. RT-PCR confirmed low levels of exon skipping after 7 days (mean 6.8% exon skipping) and very weak bands were also observed after 14 days (Fig. 3f, g). To assess the systemic efficacy of the BEPO-PNA3696 depot administration, 8–9 week-old male mdx were injected s.c. with either BEPO-PNA3696 [1.9% (w/w) loading] or vehicle formulation. Two injections of 65- to 70-μL depots were performed in a 100 mg/kg total dose group and three injections of 90-μL depots for a 200 mg/kg total dose group (n = 5). Mice with PNA depots had the same weight gain as mice with vehicle depots during the study, suggesting that there was no dose-related systemic toxicity. After 21 days, muscles and organs were harvested. Unexpectedly, and in contrast to the s.c. administration study (5 × 50 mg/kg PNA3696 semiweekly injections, Fig. 2d, e), all muscles tested (TA, Gas, Quad, Sol, and Edl) were negative for newly synthesized dystrophin protein, only very few bright staining, revertant fibers were observed (Fig. 3h). RT-PCR corroborated the immunohistochemistry findings, as no skipped bands were observed in the treated muscles (Fig. 3i).
PNA-DNA heteroduplex formulation
In another approach to improve PNA pharmacokinetics (in particular, extending circulation time as well as PNA solubility), the effect of using PNA oligonucleotide and phosphorothioate oligonucleotide heteroduplexes was tested. Phosphorothioates are routinely used as or as part of oligonucleotide antisense agents and medical drugs because of prolonged systemic circulation by binding to serum proteins [24]. In addition, hybridization to the oligonucleotide should increase the solubility of the PNA aided by the phosphates. Two 20-mer oligonucleotides were designed to either form fully complementary or only partially matched duplex [with 11 bases of the PNA and with a 9 base overhang (11bpOL)] with the PNA (Table 2). These heteroduplexes were first injected i.m. to determine in vivo activity before systemic administration (n = 4). Comparing the sections of TA treated with the PNA3696 + 100% complementary DNA and PNA3696 + 11bpOL DNA heteroduplexes, the amount of dystrophin-positive fibers was higher in the latter (Fig. 4a). This difference may be attributed to the lower duplex stability of only 11 out of 20 nucleobases, and thus a more efficient dissociation of the PNA. The amount of dystrophin-positive fibers and distribution around the injection site appeared to be similar to the TAs treated with PNA3696 alone and PNA3696+no-match control. RT-PCR results confirmed that both heteroduplexes could induce exon-skipping as skipped bands (with similar intensities) were observed (Fig. 4b). PNA3696 + 11bpOL DNA heteroduplex was selected for repeated systemic administration. Through the administration of 5 × 25 mg/kg PNA heteroduplex in a semiweekly treatment regime, dystrophin-positive fibers were observed in the TA, Gas, and Quad muscles (Fig. 4c), and skipped transcripts were detected in TA, Gas, and Quad muscles by RT-PCR (Fig. 4d). The dystrophin protein levels were too low to detect by western blotting (Fig. 4f). However, the results do not indicate any (significant) difference compared to nonformulated PNA administrated i.v. (Fig. 1a–c). Imaging analysis (Fig. 4f) likewise shows little difference between the free PNA and the PNA heteroduplexes, although a slightly higher body retention (longer circulation time) was indeed indicated for the PNA/phosphorothioate (PS)-oligonucleotide heteroduplex (as anticipated), as well as higher kidney accumulation (Fig. 4g, h).

In vivo activity of PNA-DNA heteroduplex.
The Fully Complementary, 11 Base Pair Overlap, and No-Match Control Sequences of Oligonucleotides Used for the Formation of the Peptide Nucleic Acid/Oligonucleotide and Peptide Nucleic Acid/Phosphorothioate Oligonucleotide Heteroduplexes
Discussion
In this study, we demonstrate that a 20-mer PNA can successfully be administered systemically through intravenous and subcutaneous routes. We also show that controlled release through subcutaneous depot administration of a BEPO-PNA formulation is possible, as is i.v. administration of PNA-oligonucleotide heteroduplexes. However, in none of these cases did we detect (significantly) improved antisense redirection of dystrophin mRNA splicing or increased dystrophin levels in the mdx muscular dystrophin mouse model when compared to simple i.v. administration. In vivo fluorescence imaging analysis showed (the expected) fast renal/bladder excretion of the PNA (t½ ∼ 20 min) for i.v. administration, while s.c. administration showed a two to three times slower excretion. The BEPO-PNA formulation allowed modulation of the pharmacokinetics profile as PNA release exhibited biphasic kinetics with a slow release (t½ ∼ 10 days) of 50% of the dose. In all cases, some accumulation in kidneys and liver could be detected.
Furthermore, it was not possible to dose more than 50 mg/kg i.v. due to acute toxicity, which we ascribe primarily to the need of using an acidic injection solution (pH ∼4) to avoid PNA precipitation. A slightly higher dose was tolerated by s.c. administration using a more acidic solution (pH ∼3). In addition, it should be considered that significant PNA precipitation may be expected in the vein at the injection site, when the acidic PNA solution is neutralized by the blood. Indeed, in vitro injection of the 50 mg/kg administration solution into calf serum caused significant instant precipitate formation (PNA and/or protein) (Supplementary Fig. S2). It is likely that similar precipitation/aggregation takes place upon i.v. administration, and it is uncertain whether such aggregates subsequently dissolve or may themselves cause toxic reactions. This may also explain the low efficacy in the treatment studies in mdx mice with high-dose PNA. However, the imaging data do not indicate any significant PNA accumulation at the injection site upon i.v. administration with AF680 plus a treatment dose (10, 25, and 50 mg/kg).
It is noteworthy that Gao et al. reported significantly higher activity at 3 × 50 mg/kg i.v. (dystrophin expression was ∼2% of wild-type control) [17] than we observed, and surprisingly a 20-fold higher activity using a dosing scheme of 5 × 100 mg/kg once a week for 5 weeks, yielding up to 40% of normal dystrophin expression. It is difficult to reconcile these results with our findings, and unfortunately, the description by Gao et al. concerning injection sample preparation lacks detail. It is also unclear what the purity of the PNA was (no indication of TFA content) and thus how this reported dose compares to the one we have used, which is based on pure PNA amount. Nonetheless, in our hands, it was not possible to make a 1 mM solution of the PNA (as required for dosing 50 mg/kg) in phosphate-buffered saline or another buffered medium without heavy precipitation, and in unbuffered media where the PNA was soluble (eg, isotonic glucose), the acidity dropped below pH2, and injection of such solutions is not acceptable due to toxicity. In addition, preparing a 2 mM PNA solution (as required for dosing 100 mg/kg) by the NaOH neutralization method (see Materials and Methods section) caused PNA precipitation before the pH reach 3–4.
Our attempt of hybridization of the unmodified PNA to its complementary DNA oligomer improved the solubility under neutral conditions (citrate buffer was used as a vehicle). The in vivo activity of the heteroduplex is dependent on whether the PNA can dissociate from its complementary DNA oligomer (possibly aided by oligonucleotide degradation) to bind to its target mRNA. Although the amount of dystrophin-positive fibers and percentage skipping in PNA-DNA heteroduplex-treated muscles are lower than the amount required to alleviate the phenotypic symptoms of muscular dystrophy, increased solubility under neutralized solutions was observed. However, the pharmacokinetic profiles obtained from the IVIS studies did not improve the circulation time of the heteroduplex PNA compared the AF680-PNA alone.
It is quite clear from these results that the potency of an unmodified PNA antisense agent for muscle targeting even in the case of compromised muscle fibers in muscular dystrophy is very far from promising in relationship to therapy regardless of the administration route and long-acting depot or heteroduplex formulation. Therefore, either more sophisticated formulations such as designed nanoparticles [3,4] or conjugation to cell delivery ligands (eg, cell penetrating peptides [CPPs] as used for PMOs) must be utilized to improve both pharmacokinetics as well as tissue availability. Nonetheless, the results show that in a broader context, s.c. administration of PNA may have advantages both for animal model studies as well as eventually in the clinic in terms of dosing flexibility (easier, higher, and less frequent dosing regimens), and also regarding sustained release. In addition, BEPO technology should be further explored, since these data indicate that this may be a feasible and convenient route to obtain long-term (days/weeks) sustained, systemic release. Different parameters such as polymer nature, polymer content, TB:DB ratio, and PNA loading could be adjusted to improve the pharmacokinetics profile of a BEPO-PNA formulation even further.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.