
Abstract
The development of delivery vehicles for small interfering RNAs (siRNAs) remains a bottleneck to widespread clinical use. Cationic polymers represent an important class of potential delivery vehicles. In this study, we used alkyne-azide click chemistry to synthesize a variety of cationic poly(propargyl glycolide) backbone polymers to bind and deliver siRNAs. We demonstrated control over the binding interactions of these polymers and siRNAs by varying binding strength by more than three orders of magnitude. Binding strength was found to meet or exceed that of commercially available transfection agents. Our polymers effectively delivered siRNAs with no detectable cytotoxicity. Despite accumulation of siRNAs at levels comparable with commercial reagents, we did not observe silencing of the targeted protein. The implications of our results for future siRNA delivery vehicle design are discussed.
Introduction
Widespread use of small interfering RNAs (siRNAs) in clinical practice will require improvement in the approaches and molecules used for siRNA delivery. Viral and nonviral delivery approaches are being developed. Although viral vectors are generally more efficient [1], safety concerns make further development of nonviral delivery approaches essential. To maximize specific activity of the delivered siRNA, delivery vehicles must be designed to (1) protect siRNAs from degradation by serum nucleases, (2) avoid nontargeted tissues, (3) be endocytosed efficiently by the target cells, (4) allow siRNAs to escape endosomal vesicles, and (5) be cytocompatible [2–4]. To date, most studies that have explored the structure–function relationships of siRNA delivery vehicles have used cell culture screens to identify delivery systems for further investigation in vivo [5]. The basic chemistries used in these studies are typically cationic lipids or polymers, as they can readily complex with siRNAs by electrostatic self-assembly.
Cationic polymers provide tunability in designing delivery systems with controlled physical and chemical characteristics [5,6]. The size of cationic polymer–siRNA complexes can be controlled by altering the length of the polymer, the relative amounts of hydrophobic and hydrophilic functionality of the polymer, and the amount of siRNA incorporated in the complex [7]. Charge and charge density can be controlled by varying the chemistry and quantity of cationic monomer or through incorporation of noncharged segments such as poly(ethylene glycol) (PEG) [8]. Incorporation of PEG increased the half-life of siRNA-containing complexes in vivo and generally decreased cytotoxicity [8]. In addition, monomers can be selected to add biodegradability and pH sensitivity to block co-polymeric complexes that have been proven effective siRNA delivery vehicles [9–11].
In this work, we sought to identify structure–function relationships for polymeric siRNA delivery vehicles. To achieve structural diversity, we used a postpolymerization modification strategy to synthesize a diverse set of polymers using copper(1)-catalyzed alkyne-azide cycloaddition (CuAAC), a type of “click” chemistry, to attach various side chains to a degradable poly(propargyl glycolide) (PPGL) backbone [12–15]. Using this approach allowed us to test polymers with structures/functionalities that mimic polymeric vehicles that have previously proven useful as delivery vehicles and to create and test new structures. We measured the in vitro binding of our polymers to short nucleic acids. Subsequently, we tested their utility in delivering siRNAs for silencing in cell culture. Although we were able to vary binding over a wide range (from 13 μg/mL to >10,000 μg/mL) and successfully delivered fluorescent siRNAs to cells, silencing was not achieved using any of the polymers. Possible explanations for the lack of silencing activity include a lack of release of siRNAs from the endosome, rapid recycling of siRNA complexes out of the cells, or poor delivery of siRNAs to the cells that was masked by accumulation of free fluorophore in the cells.
Materials and Methods
Materials
A detailed list of materials is provided in the Supplementary Data.
Polymer preparation
A Schlenk flask was used to dissolve 40–60 mg of PEG-co-PPGL into dimethylformamide (DMF). Development of the alkyne-functionalized PPGL and the CuAAC procedure were described previously [6]. The desired mole fractions of amine and alkyl sidechains (azide-functionalized) and 24 mole percent sodium ascorbate were then added. The flask was degassed three to four times using a freeze–pump–thaw cycle and backfilled with nitrogen gas. A 0.1 M CuCl2·2H2O solution was dripped in and stirred overnight at room temperature. The resulting solution was filtered to remove solids. Copper ions were removed by adding Amberlite IRC-748 ion exchange resin beads before filtering again. The DMF was removed in vacuo. Remaining polymer was dissolved in a 3:1 water/acetone mixture and dialyzed in a 12–14 kDa MWCO dialysis bag for 2–3 days. The dialysis solvents were removed in vacuo. Structures of sidechains are given in Supplementary Fig. S1.
Polymer binding gels
Polymers at varying concentrations were dissolved in 18 MΩ water containing 200 nM 6-carboxyfluorescein-tagged double-stranded DNA (dsDNA) and incubated for 15 min. dsDNA is less susceptible to degradation during the gel electrophoresis process than siRNA, and we have previously shown the use of dsDNA to test polymer binding to be a suitable, cost-effective replacement for siRNA while exhibiting similar binding to the synthesized polymers [16]. A 0.8% agarose gel containing 2 rows of 13 lanes was used for two, simultaneous independent experiments, to separate bound from free dsDNA. The outermost lanes contained no polymer and were used as control intensities for 100% unbound dsDNA. The fraction of bound dsDNA was calculated by dividing the intensity of the free DNA signal in each experimental lane by the intensity of the free DNA signal in the control lane and subtracting the resulting ratio from 1. Calculated fractional binding values from all independent experiments were plotted together. Data were then fit using a modified Hill equation [17] [Equation (1)], where K is the binding coefficient at 15 min, n is a fitted parameter that suggests the presence of cooperative binding, and [P] is the concentration of polymer.

The term binding coefficient is used in place of the dissociation constant in the Hill equation, because dissociation constant indicates the polymer–siRNA complexes reached equilibrium. As we only used an incubation period of 15 min before testing binding, we cannot guarantee equilibrium was reached in all cases.
Cell culture
Cells were maintained as described in Portis et al. [16]. In brief, NCI-H1299 (human lung carcinoma) cells expressing enhanced green fluorescent protein (EGFP) with 2-h half-life were obtained from Dr. Jørgen Kjems, University of Aarhus, Denmark [18]. Cells were passaged approximately weekly by trypsinization. Cells were grown in Dulbecco's modified Eagle's medium high glucose supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% geneticin and incubated at 37°C, 5% CO2, 100% relative humidity.
EGFP silencing and cytotoxicity
NCI-H1299-EGFP cells were plated at 20,000 cells per well in 0.1 mL of complete media without antibiotics in a 96-well black side, clear bottom plate and incubated for 24 h. Twenty-four microliters of Opti-Mem was used to complex siRNA and polymer for final concentrations of up to 100 nM siRNA and sufficient polymer to bind the siRNA according to the results of the binding gels. These were mixed and incubated at room temperature for 30 min before transfection. Linear poly(ethylenimine) (LPEI)– and Lipofectamine 2000 (LF2K)–siRNA complexes were prepared to confirm the activity of the siRNA. Transfection solution was left on the cells for 24 h at 37°C, 5% CO2, and 100% humidity. Before measuring EGFP fluorescence using a Gemini EM fluorescent plate reader (480 nm excitation/525 emission), cells were washed twice with DPBS. Cytotoxicity was determined by comparing the average EGFP signal from 3 wells of a 96-well plate of NCI-H1299-EGFP cells treated with only transfection media with the average EGFP signal from 3 wells treated with transfection media containing polymer. Silencing was measured by comparing the average EGFP signal from 3 wells of a 96-well plate of NCI-H1299-EGFP cells treated with transfection media containing polymer–siRNA complexes with the average EGFP signal from three wells treated with transfection media containing only polymer (no siRNA).
Microscopy
Transfection media was removed, and cells were rinsed before being placed in 0.5 mL of Leibovitz Medium (L-15) media for imaging. An Olympus FluoView 1000 Inverted IX81 microscope with 40 × oil objective was used for confocal laser scanning microscopy. EGFP was excited using a 488 nm multi-line Argon laser and detected through a BA505-52 nm emission filter. Dy547-tagged siRNA was excited with a 543 nm HeNe laser and detected through a BA560-IF nm filter. Imaging was performed sequentially using a Kalman average of 2 at the focal plane with the highest intensity EGFP signal.
Results
By using click chemistry to modify PEG-PPGL backbone polymers, we were able to rapidly synthesize 59 unique cationic biodegradable polymers to be complexed with siRNA for transfection of NCI-H1299 cells. The polymers consisted of four structural elements (Fig. 1A). PEG and PGL were linked and served as the polymer backbone. “Click” chemistry (Fig. 1B) was used to functionalize the backbone with amine-containing or alkyl sidechains (Fig. 1C). PEG length was varied, with each polymer having a PEG segment length of 0, 8, or 115 repeat units (Table 1). PGL length was also varied from 5 to 150 repeat units. The amine functional groups added to the PGL backbone were either primary, secondary, tertiary, or a combination of primary and secondary amines (Fig. 1C). Alkyl sidechains ranged from 4 to 16 carbons in length.
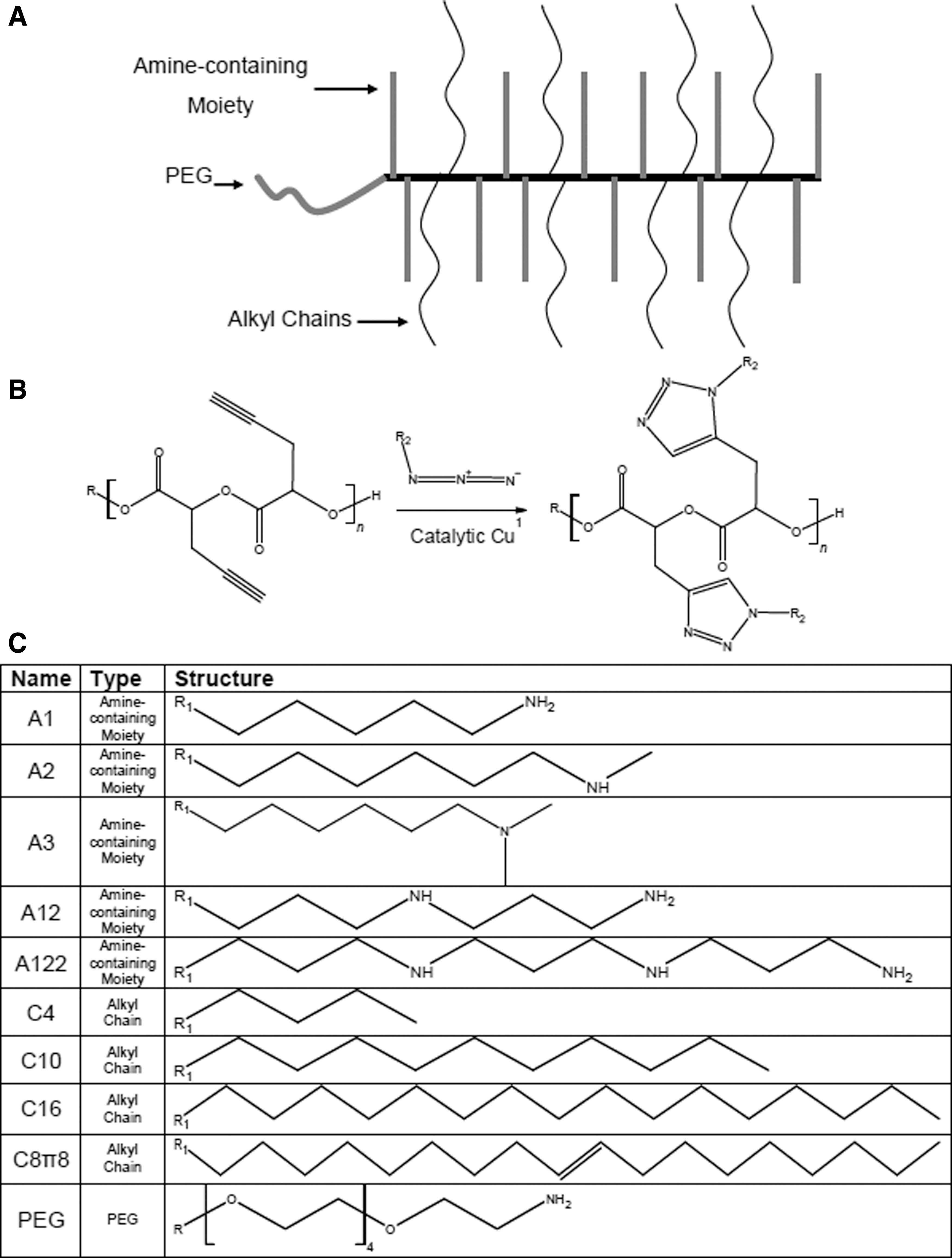
Synthesis and polymer schematic.
Name, Structure, and Binding Properties of Vehicles Used for Small Interfering RNA Delivery
BPEI, branched poly(ethylenimine); LF2K, Lipofectamine 2000; LPEI, linear poly(ethylenimine); PEG, poly(ethylene glycol); PLL, poly-
Polymer–nucleic acid complex formation was measured using a gel-shift assay (Fig. 2). Binding coefficients, K, varied from 13 μg/mL to >10,000 μg/mL (Table 1). We also tested binding for a number of commercially available siRNA delivery systems that have demonstrated silencing [poly-
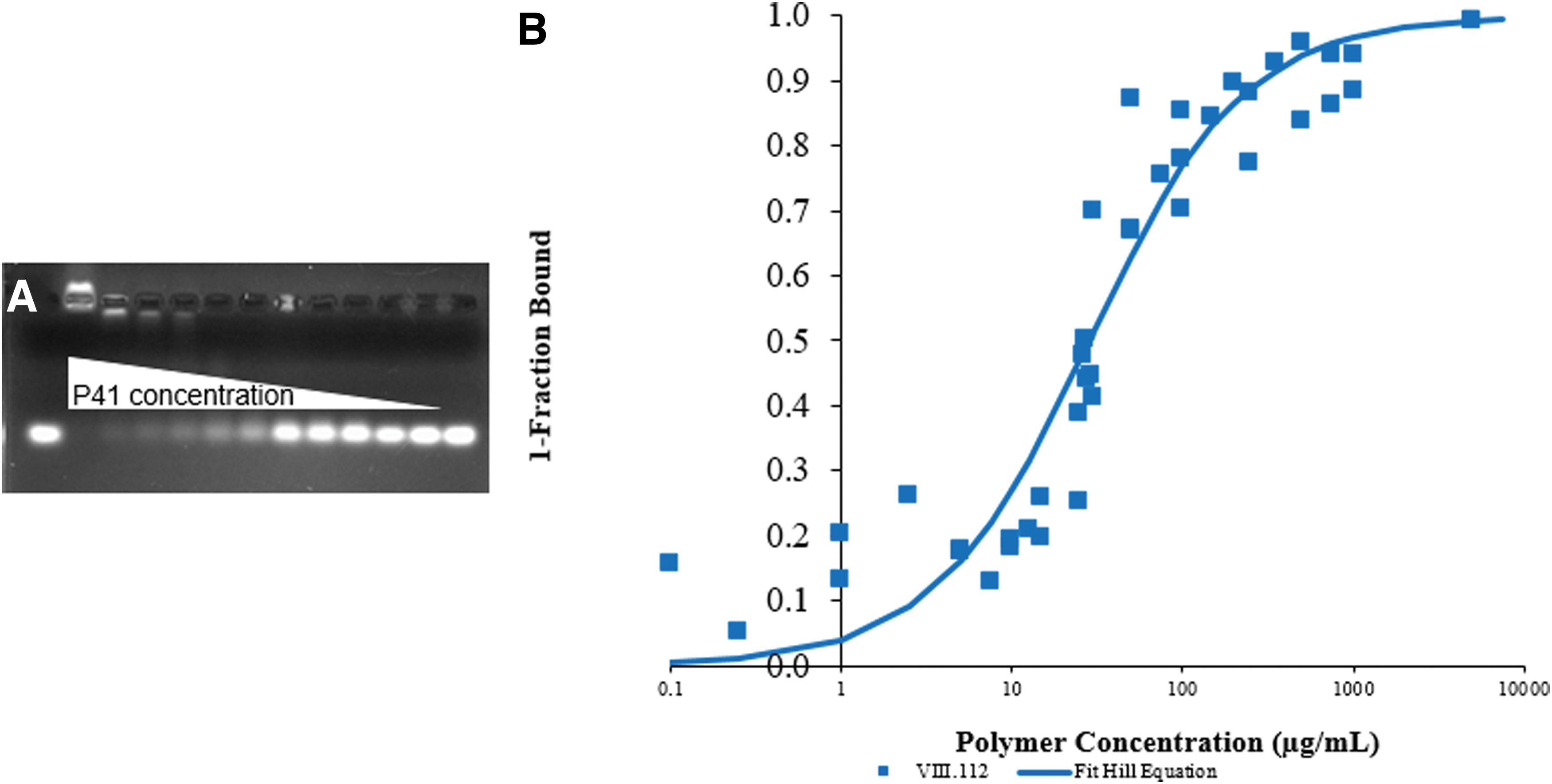
Gel-shift Analyses.
Confocal laser scanning microscopy was used to test the ability of our polymers to deliver siRNAs. A representative example, polymer P3, delivered siRNA to NCI-H1299 cells (Fig. 3); however, despite accumulation at levels comparable with those achieved with LF2K (Fig. 3, compare C and I), the siRNAs did not reduce the expression of EGFP. Indeed, none of the synthesized polymers led to a statistically significant decrease in EGFP, regardless of the strength of the binding interaction between the siRNA and polymer.
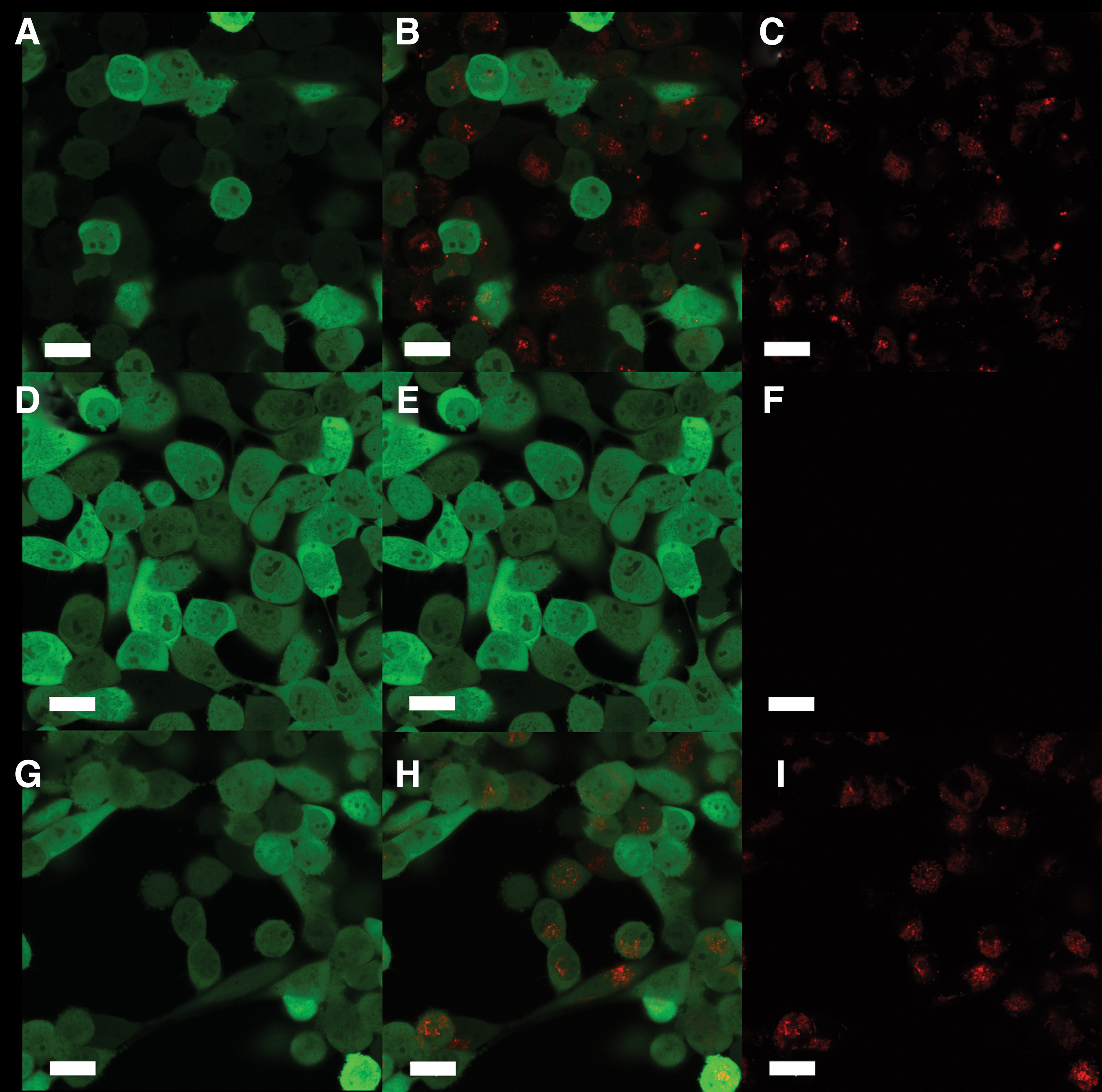
siRNA accumulation and silencing. Shown are the delivery of Dy547-siRNA and silencing of EGFP in NCI-H1299-EGFP cells, all scale bars are 20 μm.
Discussion
For siRNAs to accumulate in cells, delivery vehicles must bind them and protect them from nuclease degradation during delivery. We have shown increasing PEGylation of polymers impairs their binding with dsDNA/siRNA, whereas combining primary and secondary amines, increasing the number of amines, and increasing the length of alkyl sidechains improves binding. It has been shown previously that PEGylation decreases binding affinities of nucleic acid/polymer complexes by charge shielding [8]. Of interest, isolated primary, secondary, and tertiary amines all have pKa values above physiological pH; therefore, differences in pKa among the amine classes do not account for the differences in binding. The inclusion of two nearby amines in the A12 sidechain potentially allowed for multiple binding sites between the siRNA and polymer, stabilizing the electrostatic interaction [19].
Binding of a polymer to siRNAs can be sterically limited [20]. Incorporation of alkyl chains can produce nanoparticles with a hydrophobic core with cationic arms extending into the hydrophilic solution [21]. These arms are then more spatially available to conform to the siRNA, improving binding. Amphiphilic polymer nanoparticles can also form micelle-like conformations surrounding siRNAs [8]. The inclusion of a double bond in P10 impairs binding compared with P5 with the same number of carbons (Table 1), indicating that hydrophobic packing may be crucial for the enhanced binding observed with longer alkyl sidechains. This further supports our hypothesis that these polymers form a micellar structure.
It is notable that many of our polymers had binding interactions of similar strength to those of the commercial vehicles. Moreover, our polymers successfully delivered siRNAs into cells at comparable concentrations. Nonetheless, none of the polymers tested was able to lead to silencing of EGFP. One possible explanation for the lack of efficacy of the polymers is the absence of secondary and tertiary amines, which can buffer solutions as low as pH = 3 and therefore may lead to the proton sponge effect and endosomal rupturing to release siRNAs [22,23]. Amphiphilic siRNA delivery vehicles have also been shown to disrupt endosomal membrane stability and thereby allow siRNAs to enter the cytoplasm [24]. To disrupt the membrane, delivery vehicles must be near neutral charge at endosomal pH to avoid electrostatic exclusion from the membrane [24]. However, our polymers are likely too positively charged to interact with the membrane in this manner. Another possible explanation for the lack of effective delivery by our polymers is that endosomes containing the polymers are quickly tagged for recycling or degradation, not allowing for sufficient time to release their cargo [25]. We have previously shown that accumulation of siRNAs in cells does not directly correlate to the degree of silencing achieved [26]. Thus, it is possible that the siRNAs delivered by our polymers may have been endocytosed for recycling but precluded from reaching the cytoplasm and initiating RNAi. Finally, it is possible that the Dy547 fluorophore detaches from the siRNA before cellular internalization and enters either alone or with the PPGL nanoparticle. It is unclear why this would be more likely to occur during delivery by the polymers than by LF2K, but, if it were the case, our levels of delivered siRNA may be insufficient to achieve silencing.
The development of effective and safe delivery systems remains a limiting step in the clinical application of siRNAs. Our polymers offer a unique opportunity to assess the vehicle characteristics that influence downstream siRNA activity because of their facile modification by click chemistry. The benefits of having multiple classes of amines incorporated into one polymer remain unclear but could contribute to better binding affinity or an increased buffering capacity. The incorporation of higher molecular weight alkyl sidechains requires future investigation to find the length of alkyl chain that optimizes binding with siRNA without impairing solubility or increasing size. Finally, it will be important to understand how alkyl sidechain length impacts stability, conformation, and, ultimately, the delivery effectiveness of polymeric vehicles for siRNAs.
Footnotes
Acknowledgments
The authors thank the members of the Cellular and Biomolecular Laboratory for their advice and support. The authors acknowledge the contributions to this work of the late Gregory L. Baker, our mentor, colleague, and friend, who was an exceptional polymer scientist and instrumental in the initial design and execution of this collaborative work.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Michigan State University, the National Science Foundation [CBET1510895, 1547518, 1802992], and the National Institutes of Health [GM089866, T32GM092715 to R.C.S.].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.