
Abstract
A series of 2′-deoxy and novel 2′-O-methyl and 2′-O-(2-methoxyethyl) (2′-MOE) oligonucleotides with internucleotide methanesulfonyl (mesyl, μ) or 1-butanesulfonyl (busyl, β) phosphoramidate groups has been synthesized for evaluation as potential splice-switching oligonucleotides. Evaluation of their splice-switching activity in spinal muscular atrophy patient-derived fibroblasts revealed no significant difference in splice-switching efficacy between 2′-MOE mesyl oligonucleotide and the corresponding phosphorothioate (nusinersen). Yet, a survival study with model neonatal mice has shown the antisense 2′-MOE mesyl oligonucleotide to be inferior to nusinersen at the highest dose of 40 mg/kg. A reason for their lower activity in vivo as ascertained by cellular uptake study by fluorescent confocal microscopy in HEK293 cell line could possibly be ascribed to compromised endosomal release and/or nuclear uptake of the 2′-OMe or 2′-MOE μ- and β-oligonucleotides compared to their phosphorothioate analog.
Introduction
Antisense oligonucleotides have been extensively reviewed in the literature as therapeutic agents that target biologically important RNAs through either RNase H mediated enzymatic cleavage [1–3] or steric modulation of RNA metabolism [4,5]. While the former type is applicable to a wider range of potential targets, including genetic diseases [6], cancers [7], and viral [8] and bacterial [9] pathogens, where the target RNA can be destroyed, the latter type works when subtler changes in the RNA activity are required such as splice redirection involving exon skipping or inclusion [10,11]. A primary therapeutic target in the latter case are monogenic genetic diseases such as Duchenne muscular dystrophy [12] and spinal muscular atrophy (SMA) [13]. Up to date, several steric blocking antisense splice-switching oligonucleotides have been approved by U.S. Food and Drug Administration (FDA) for clinical use such as eteplirsen (Exondys 51) [14], golodirsen (Vyondys 53) [15], and nusinersen (Spinraza) [16]. Among these, nusinersen has received public recognition as a ‘life-saving drug’ to treat SMA.
SMA is a leading genetic cause of infant mortality primarily due to motor neuron degeneration and progressive muscle weakness [17]. It results from loss of the ubiquitous survival motor neuron 1 gene, SMN1 [17,18]. Human genome contains a second nearly identical copy, SMN2, which differs from SMN1 by a crucial nucleotide transition within exon 7 leading to the predominant generation of an alternative exon 7-excluded mRNA transcript and only marginally functional protein [19–23]. SMN2 therefore fails to compensate for loss of SMN1 unless sufficient copies are present to generate functional levels of full-length SMN protein [18].
A therapeutic approach for SMA utilizes single-stranded antisense splice-switching oligonucleotides to promote inclusion of exon 7 into SMN2 pre-mRNA through steric blocking of pre-mRNA elements pertinent to splice regulation [24]. The clinically approved antisense oligonucleotide against SMA, nusinersen (Spinraza), is an 18-mer oligo-2′-O-(2-methoxyethyl) (2′-MOE) ribonucleotide (Fig. 1) that targets the intron splice silencer N1 (ISS-N1) site within intron 7 [25,26]. Nusinersen improves survival and motor function in the majority of SMA patients, most prominently in those treated early in disease progression [27]. The drug is administered through intrathecal injection, which to-date appears to be reasonably safe and tolerable in both adult and children patients, even those with spinal problems [28,29].
Backbones for SSOs: oligo-2′-MOE-ribonucleotide phosphorothioate (2′-MOE PS) (
The backbone of nusinersen is the phosphorothioate: a phosphate linkage isostere, which is present in a majority of antisense oligonucleotides studied so far [30]. Phosphorothioate groups (PS) increase enzymatic stability and propensity for protein binding that improve cellular uptake, biodistribution, and pharmacokinetic properties of the oligonucleotides (PS ONs) [31,32]. However, adverse effects of PS ONs were also documented, most notably, in vivo toxicity, particularly liver damage [33,34] and complement activation [35,36]. The concerns over clinical safety of PS ONs have resulted in the repeated refusals of European Medicines Agency to approve mipomersen (Kynamro), an FDA-approved drug to treat homozygous familial hypercholesterolemia [37], and failure of several PS-modified drug candidates to make through different phases of clinical trials [38,39]. Although it was recently reported that a pinpoint sugar modification improved the therapeutic index of RNase H-competent PS ONs [40], the adverse effects associated with the use of phosphorothioate group for antisense ONs remain a serious concern, especially for splice-switching applications, where the need for a life-long treatment was postulated [41]. This highlights the importance of devising new chemistries for antisense oligonucleotide backbone modification.
Recently, we have shown that mesyl (methanesulfonyl) phosphoramidate group (μ) can be an efficient alternative to phosphorothioate (PS) group in antisense oligonucleotides targeting miRNAs [42]. The corresponding RNase H-competent mesyl phosphoramidate oligodeoxynucleotides (μ-ODNs) (Figs. 1 and 2A) were demonstrated to possess increased specificity, reduced toxicity, and improved activity over their PS analogs [42]. The results encouraged us to attempt the design of novel steric blocking oligonucleotides incorporating mesyl phosphoramidate (μ) or their extended chain analog 1-butanesulfonyl (busyl) phosphoramidate (β) groups [43] as potential splice-switching agents by replacing 2′-deoxynucleosides in the backbone with 2′-O-methylribonucleosides (Figs. 1 and 2B) or 2′-MOE ribonucleosides (Figs. 1 and 2C). Such sugar modifications are known to increase stability of antisense oligonucleotide duplexes with RNA and completely abolish RNase H activation [44], which is essential for splice-switching applications. We would like to present herein our studies on the cellular uptake and intracellular localization of 2′-MOE mesyl phosphoramidate oligonucleotides (MOE μ-ONs) and a range of 2′-substituted mesyl and busyl analogs, and evaluation of their splice-switching activity in SMA patient-derived fibroblasts and in vivo in a neonatal mouse model of SMA.
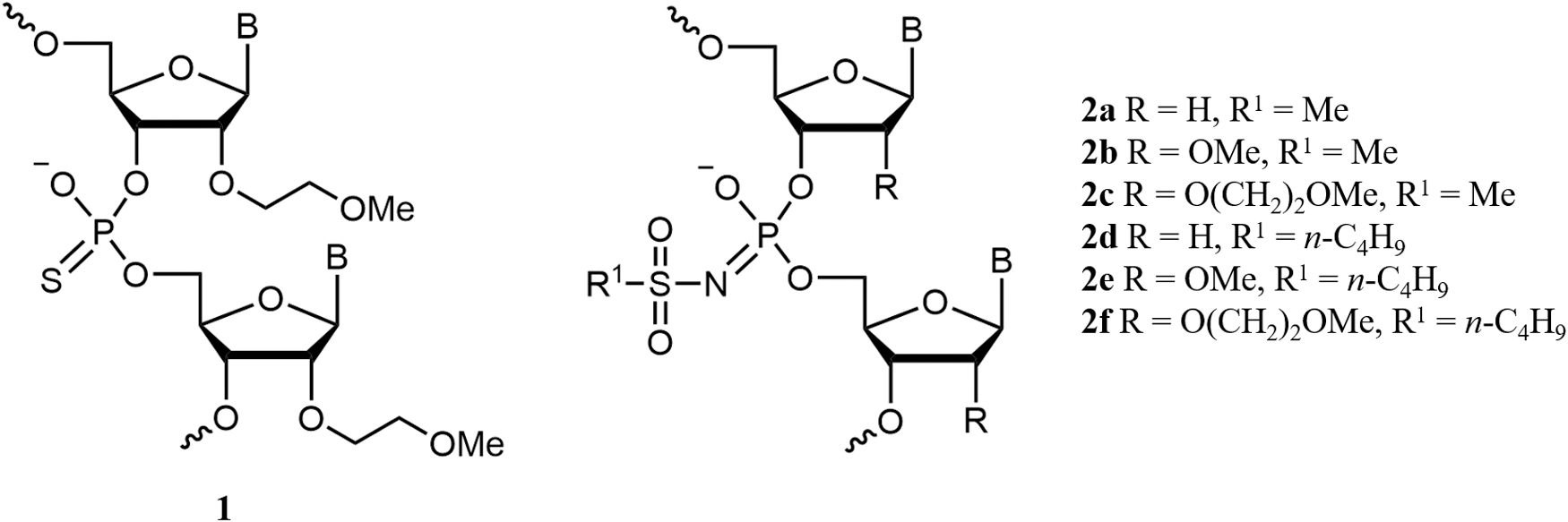
Splice-switching activity of ASOs in SMA patient fibroblasts. Cells were treated with 0, 1, 10, or 50 nM ASOs by lipofectamine transfection agent. Splice-switching activity was analyzed by qPCR (mean ± SEM). ASO chemistries
Materials and Methods
Oligonucleotide synthesis
Mesyl (μ) and busyl (β) phosphoramidate oligonucleotides (Table 1) were assembled by automated solid-phase phosphoramidite synthesis replacing aqueous iodine oxidation by Staudinger reaction with either methanesulfonyl azide or 1-butanesulfonyl azide (0.5 M in acetonitrile, 40 min at ambient temperature) in each cycle of chain elongation [43,45]. After the completion of the synthesis, the oligonucleotides were cleaved from the polymer support and deprotected by standard ammonia treatment followed by consecutive PAGE and reverse-phased (RP) high performance liquid chromatography (HPLC) purification as described previously [46]. For confocal microscopy studies, 3'-alkynylated mesyl and busyl phosphoramidate oligonucleotides were labeled with Cy5 azide using Cu(I)-catalyzed 1,3-dipolar alkyne-azide cycloaddition reaction (CuAAC) [46]. As PS oligonucleotides have been reported to undergo significant desulfurization under Cu(I)-mediated click reaction [47], N-hydroxysuccinimidyl (NHS) ester of Cy5 dye has been used for acylation of 3'-aminoalkylated oligonucleotide in the latter case. Molecular masses of the oligonucleotides were verified by ESI LC-MS (Table 1).
Sequences and Molecular Masses of Modified Oligonucleotides
Conditions of ESI-MS: see Supplementary Data.
(S)—phosphorothioate group.
Cy5 fluorescent label.
(μ)—mesyl (methanesulfonyl) phosphoramidate group.
(β)—busyl (1-butanesulfonyl) phosphoramidate group.
Cm—5-Me-C residue.
2′-MOE, 2′-O-(2-methoxyethyl).
In vitro evaluation of splice-switching activity in SMA patient fibroblasts
Oligonucleotides were resuspended in nuclease-free water. Type II SMA patient-derived fibroblasts (GM03813; Coriell Institute for Medical Research) were maintained in standard medium (DMEM+GlutaMax (cat. No. 31966-021; Thermo Fischer Scientific), 15% fetal bovine serum (FBS), and 1% penicillin-streptomycin). Forty-eight hours before treatment, fibroblasts were seeded in 6-well plates at the density of 140,000 ¢/well.
Lipofectamine 3000 (cat. No. L3000-001; Invitrogen)-assisted transfection experiments were performed in 15% FBS, according to the manufacturers' manual. All conditions were conducted in technical triplicates.
After 24 or 72 h post-treatment, cells were washed with phosphate-buffered saline (PBS), and cell pellets were collected after trypsinization and centrifugation (500 g, 5 min, at 4°C).
RNA was extracted using the Promega Maxwell® RSC simply RNA Cells kit (cat. No. AS1390; Promega), and eluted in 35 μL nuclease-free water. RNA concentration was measured using an ultraviolet (UV)-Vis spectrophotometer Nanodrop 2000 (ThermoFischer Scientific).
cDNA was prepared from 1 μg RNA, using ABI High-Capacity cDNA Reverse Transcription Kits (cat. No. 4368813; Applied Biosystems), following the manufacturers' instructions, and was then diluted to a final concentration of 10 ng/μL.
Quantitative PCR (qPCR) was performed on StepOne™ Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) using TaqMan™ Fast Advanced Master Mix, and primers for FL-SMN and tot-SMN as described in Ref [13]. Efficiency of the PCR was corrected for using the LinReg PCR software [48,49]. Final data were analyzed using the method described in Ref [50]. Statistical analyses were conducted with Prism v.8, GraphPad Software, which was also used to plot the figures.
Analysis of Cy5-labeled oligonucleotide nuclear uptake by confocal microscopy in fixed HEK293 cells
Solutions of Cy5-labeled antisense oligonucleotide (ASO) (Table 1) in 1 × PBS buffer were added to human HEK293 (ATCC CRL-3216) cells in Dulbecco's modified Eagle medium nutrient mixture F-12 (DMEM/F12) supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin cultured on poly-L-lysine-coated cover glass slides placed to the bottom of a 24-well plate to the final concentration of 1 μM. Cells were cultured at 37°C in humidified air with 5% CO2 for 24 h. Then, glasses with cells were washed with 1 × PBS, fixed in 1 × PBS containing 4% formaldehyde at room temperature for 20 min, washed with 1 × PBS, and permeabilized in 1 × PBS with 0.1% Triton X-100 for 5 min. Then glasses were washed twice with 1 × PBS for 10 min. Cells were embedded in Mowiol with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma) before imaging. Confocal microscopy was performed using Nikon A1+MP confocal imaging system using a Plan Apo 20x/0,75 Dic N objective (numerical aperture 0.75; Nikon Japan), Apo LWD 40x/1,15 S water immersion objective (numerical aperture 0.15; Nikon, Japan), and Apo tirf 60x/1,49 interference contrast imaging (DIC) oil immersion objective (numerical aperture 0.49; Nikon, Japan). Images were scanned sequentially using 561 nm diode lasers in combination with a DM561 nm dichroic beam splitter. To describe the nuclear membrane contour, each Z-stack was processed by the intensity threshold of the blue channel (cell nucleus) in the first stage of calculations. Thresholding was performed by the following parameters: area, roundness, and intensity of fluorescence. Next, the binary layer of nuclei was subtracted from the binary layer of the sample, allowing to detect fluorescence intensity of the ASO inside the nuclei only.
Live HEK293 cell imaging of Cy5-labeled oligonucleotides internalization by confocal microscopy
Approximately 1 × 105 HEK293 (ATCC CRL-3216) cells in DMEM/F12 supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin cultured in the confocal dishes for 24 h up to achieving 70% confluence. LysoTracker red DND-99 (Invitrogen) was added to the cells to a final concentration of 50 nM and incubated for 10 min. Then, cells were washed 3 times with 1 × PBS, and the fresh DMEM/F12 medium was added. ASOs (Table 1) in 1 × PBS were added to the cells to a final concentration of 0.1 mM and incubated for 60 min. The cells were washed 3 times with 1 × PBS, and the fresh DMEM/F12 medium was added. Live cell imaging of ASO internalization was carried out at 37°C in humidified air with 5% CO2 on a Nikon A1+MP confocal imaging system using a Plan Apo 20x/0,75 Dic N objective (numerical aperture 0.75), Apo LWD 40x/1,15 S water immersion objective (numerical aperture 1.15, Nikon, Japan). Images were scanned sequentially using 561 and 647 nm diode lasers in combination with a DM405/488/561/633 nm dichroic beam splitter. Differential DIC microscopy was used to visualize cell contours. The images were analyzed with NIS elements AR (Nikon Japan). PCC (Pearson's correlation coefficient) and MOC (Manders' overlap coefficient) were used to assess the co-localization rate.
Animal model and dosing
Animal studies were carried out in the Biomedical Sciences Unit, University of Oxford, according to procedures authorized and approved by the University of Oxford ethics committee and UK Home Office (project license PDFEDC6F0). SMA-like mouse strain FVB.Cg-Smn1tm1HungTg(SMN2)2Hung/J was generated and maintained as previously described [51,52]. 2′-MOE oligonucleotides 5'-TCA CTT TCA TAA TGC TGG targeting the SMN2 ISSN1 sequence with phosphorothioate or mesyl phosphoramidate internucleotidic linkages (MOE-Nus-s and MOE-Nus-m, respectively) were tested. Subcutaneous administrations were performed by single (PND0) administrations. Doses of 20 μg/g (2.35 nmol/g for MOE-Nus-m or 2.73 nmol/g for MOE-Nus-s), 30 μg/g (3.58 nmol/g for MOE-Nus-m or 4.10 nmol/g for MOE-Nus-s), or 40 μg/g (4.70 nmol/g for MOE-Nus-m or 5.47 nmol/g for MOE-Nus-s) of ASOs were diluted in 0.9% saline and given at a volume of 10 μL per gram body weight. Scrambled mesyl and phosphorothioate oligonucleotides MOE-Scr-m and MOE-Scr-s 5'-ACT TTC ACT AAT CGT GGT and luciferase-targeted MOE-Luc-m and MOE-Luc-s 5'-TCG AAG TAC TCA GCG TAA G were used as controls. Treated mice were given mash as food supplementation. Weights were taken daily and overall health assessed. Following the humane endpoint, mice were euthanized according to the approved procedure of rising CO2.
Results
Design and synthesis of novel mesyl and busyl phosphoramidate oligo-2′-O-alkyl ribonucleotides
The synthesis of 2′-O-alkyl mesyl (μ) and busyl (β) oligonucleotides (Figs. 1 and 2) was accomplished according to our previously reported method of automated solid-phase assembly through the phosphoramidite method followed by Staudinger reaction with 0.5 M solution of a sulfonyl azide in acetonitrile instead of aqueous iodine oxidation [43,45]. The Staudinger reaction between the support-bound phosphite triester formed during the phosphoramidite coupling and methanesulfonyl azide or 1-butanesulfonyl azide, respectively, was carried out for 40 min at ambient temperature. Oligonucleotides containing 5'-dimethoxytrityl group (DMTr) were isolated by RP HPLC or, if necessary, purified to homogeneity by denaturing PAGE under the same conditions as phosphodiester or PS ONs followed by RP-HPLC to remove phosphodiester impurities. The molecular masses of the μ- and β-oligonucleotides obtained have been confirmed by ESI LC-MS (Table 1).
In vitro evaluation of splice-switching activity in SMA patient fibroblasts
Splice-switching activity of ASOs in vitro has been assessed in SMA patient-derived fibroblasts. Due to low ASO uptake in fibroblasts through gymnosis, we used lipofectamine transfection agent for cell culture studies. Fibroblasts were treated with 0, 1, 10, and 50 nM SMN2-targeted ASOs with nusinersen sequence (Table 1). The nusinersen ASO sequence acts by binding to the ISSN1 sequence of SMN2 intron 7, masking a splicing suppressor region and thereby enhancing exon 7 included transcripts. Splice-switching activity was measured by qPCR relating the amount of exon 7 included transcripts (FLSMN) to the total number of transcripts (TotalSMN). TotalSMN was determined by exons 1 and 2a present in all SMN2-derived transcripts. Regardless of the phosphate modification, 2′-deoxy ASOs were not active in these cells (Fig. 2A) as it may be expected because of the different mechanism of action (RNase H recruitment). Nor did busyl phosphoramidate (β) ASOs (me Nus-bm and MOE-Nus-bm) show activity, possibly hinting at unfavorable intracellular localization of β-ASOs. Active ASOs were the 2′-OMe and 2′-MOE with phosphorothioate (PS) and mesyl phosphoramidate (μ) internucleotidic linkages (MOE-Nus-s, MOE-Nus-m, me Nus-s, and me Nus-m, respectively) (Fig. 2B, C). Scrambled ASO sequence with the most active 2′-MOE chemistry was inactive with all phosphate modifications (Fig. 2D). The most active in cell culture compounds, MOE-Nus-m and MOE-Nus-s, were selected for further comparison in vivo.
Intracellular distribution and endosomal release of phosphorothioate, mesyl (μ) and busyl (β) phosphoramidate oligonucleotides in live cells
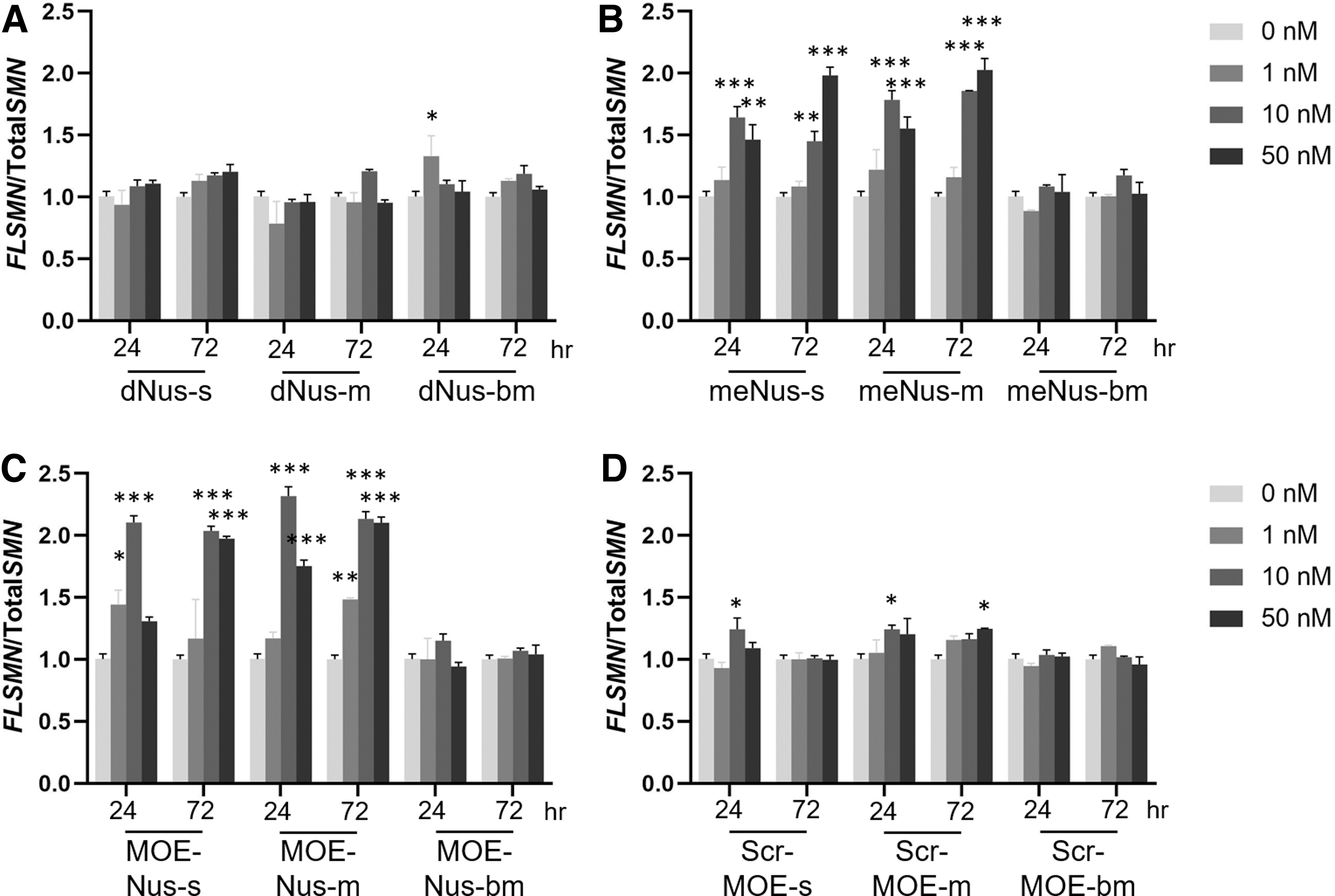
We studied intracellular distribution of Cy5-labeled ASOs in live HEK293 cells after unaided uptake (gymnosis) at 0.1 μM concentration of each ASO in the presence of fluorescent-labeled lysosome tracker (Fig. 3). Three modified phosphate derivatives, namely, phosphorothioate (PS), mesyl (μ), and busyl (β) phosphoramidate, were compared for three backbones: 2′-deoxy, 2′-OMe, and 2′-MOE. Deoxy oligonucleotides with phosphorothioate (dNus-s), mesyl (μ) phosphoramidate (dNus-m), or busyl (β) phosphoramidate (dNus-bm) internucleotidic linkages demonstrated approximately seventy-five percent of the endosomal release during the first hour of incubation based on the PCC calculation (Supplementary Fig. S2). Introduction of 2′-OMe modifications into the phosphorothioate oligonucleotide (me Nus-s) decreased the release rate of the ASO from endosomes to ∼60%. However, switching to 2′-OMe backbone in μ-ASO (me Nus-m) or β-ASO (me Nus-bm) improved endosomal release (∼83% and ∼72%, respectively), and only 17% and 28% of μ-ASO or β-ASO were found in lysosomes. 2′-O-Methoxyethyl phosphorothioate ASO (MOE Nus-s) demonstrated reduced release (∼65% only). In the case of 2′-MOE mesyl phosphoramidate (MOE Nus-m), the increase of accumulation of μ-ASO in the lysosomes was up to ∼60% (∼40% of release of μ-ASO in the cell). At the same time 2′-MOE busyl phosphoramidate (MOE Nus-bm) was released from endosomes up to ∼72% under the same conditions (Supplementary Fig. S2). Thus, increased hydrophobicity of the μ-ASO or β-ASO improves endosomal escape only in the case of 2′-O-alkyl RNA backbone.

Confocal microscopy images of ASO internalization after 60 min in live HEK293 cells with labeled lysosomes.
Intranuclear distribution of phosphorothioate, and mesyl and busyl phosphoramidate oligonucleotides in fixed cells
We studied intranuclear distribution of Cy5-labeled ASOs in fixed HEK293 cells after unaided uptake (gymnosis) at 1 μM concentration of each ASO and 24-h exposition (Fig. 4). Confocal micrographs demonstrate that the majority of deoxy phosphorothioate (d-Nus-s) is uniformly distributed across cytoplasm and the nucleus, while some amount is concentrated in perinuclear structures. Switch from phosphorothioate to mesyl phosphoramidate in the deoxy series (d-Nus-m) resulted in ASO accumulation predominantly in the cytosol, including perinuclear space. Busyl phosphoramidate (d-Nus-bm) modification also resulted in the predominantly cytosolic accumulation of the ASO. Introduction of 2′-OMe groups into the phosphorothioate oligonucleotide (me-Nus-s) resulted in cytoplasmic localization of ASO in perinuclear structures. At the same time, 2′-OMe mesyl phosphoramidate ASO (me-Nus-m) demonstrated not only cytoplasmic localization but also some fluorescence in the nucleus. Switch to busyl phosphoramidate (me-Nus-bm) shifts the localization of ASO to perinuclear region of cytoplasm with clustering near the nucleus. 2′-MOE modification in conjunction with phosphorothioate backbone (MOE-Nus-s) caused accumulation of ASO in the perinuclear region of the cytosol with some fluorescent signals observed in the cell nucleus. Introduction of 2′-MOE modification into mesyl (MOE-Nus-m) or busyl (MOE-Nus-bm) phosphoramidate oligonucleotides resulted in their distribution to endosomes in the cytosol and clustering in the perinuclear region and nucleus.
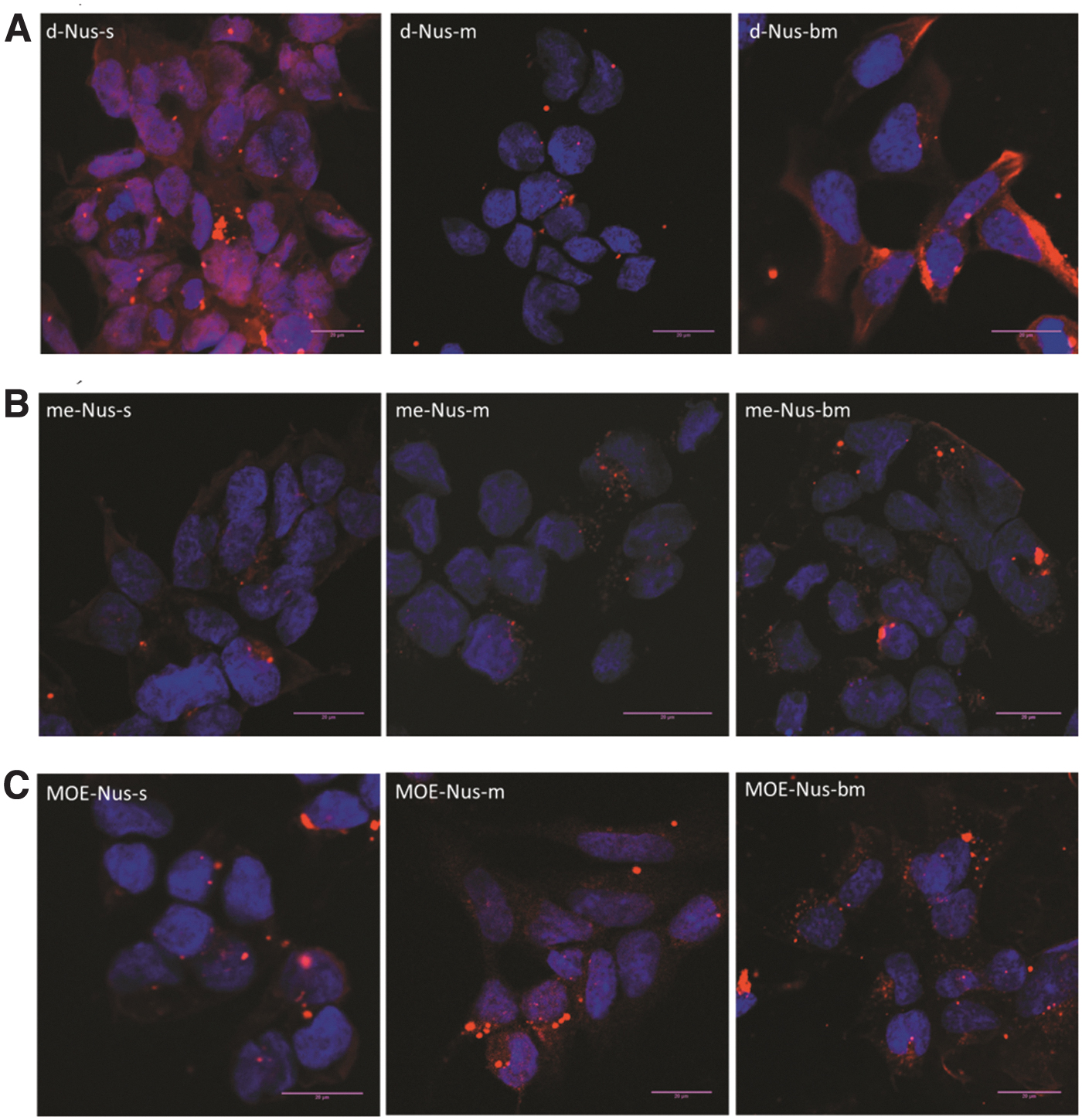
Confocal microscopy images (z-stacks) of ASO internalization after 24 h in fixed HEK293 cells.
Calculation of the fluorescent signal intensity in the cell nucleus (Supplementary Table S4) demonstrated that deoxy phosphorothioate (dNus-s) was taken to the nucleus with the highest efficacy, whereas mesyl (μ) and busyl (β) phosphoramidate modifications decreased the efficacy ∼7 fold. Introduction of 2′-OMe groups into all types of backbone modification decreased the nuclear localization by ∼3 to 4 times compared to phosphorothioate oligodeoxynucleotide (dNus-s). 2′-MOE modification demonstrated the most significant impact on the level of the intranuclear localization. 2′-MOE modification in conjunction with phosphorothioate backbone demonstrated the lowest signal in the cell nucleus. Thus, addition of the mesyl and busyl phosphoramidate modifications provides the second modality to influence on the intracellular distribution of 2′-O-alkyl oligonucleotides.
Systemic administration of 2′-MOE mesyl phosphoramidate oligonucleotide does not improve in vivo efficacy over 2′-MOE phosphorothioate (nusinersen)
The Taiwanese SMA model [51] recapitulates the severe form of SMA through significant weight loss, reduced movement, and early-onset death between PND7 and PND12. Pups were treated with 20, 30, or 40 μg/g of ISSN1-targeted oligonucleotides MOE-Nus-m and MOE-Nus-s and the respective control oligonucleotides through subcutaneous injections on the day of birth (Supplementary Tables S1 and S2, Supplementary Fig. S1). We used low doses to distinguish activity between the two ASO chemistries. Weights and survival were monitored daily (Fig. 5). Pups treated with 20, 30, or 40 μg/g MOE-Nus-s survived a median age of 9, 9, and 222 days, respectively (Fig. 5A, Supplementary Table S3). The median survival for similarly MOE-Nus-m-treated pups was 5.5, 15, and 18 days, respectively (Fig. 5B, Supplementary Table S3). Forty micrograms per gram MOE-Nus-s survival was statistically significant from untreated (P < 0.0005) as well as 40 μg/g MOE-Nus-m-treated pups (P < 0.01) (Fig. 5C). Weights between 40 μg/g MOE-Nus-s- and control MOE-s-treated pups were statistically different (P < 0.005) (Fig. 5E); however, there was no significant difference in weights at 40 μg/g dose between MOE-Nus-m and MOE-Nus-s (Fig. 5F).
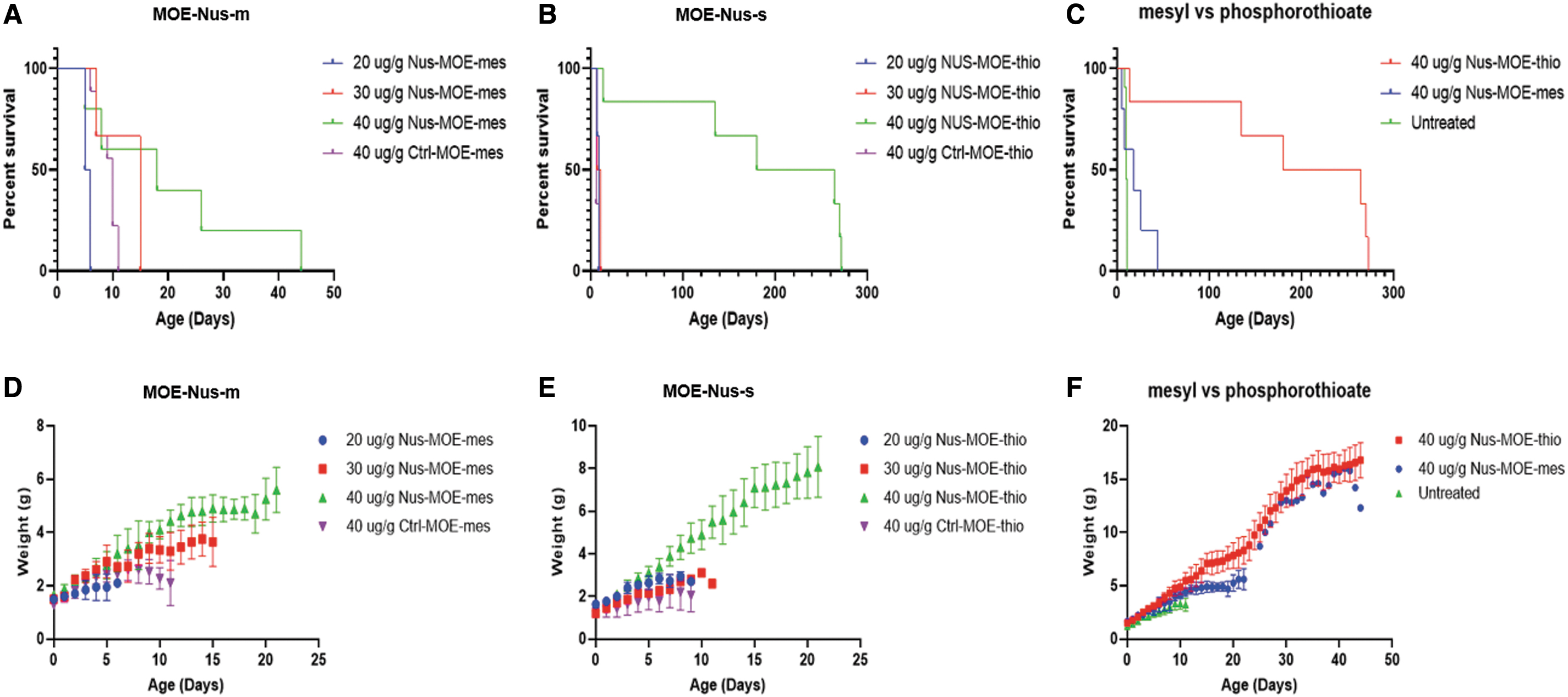
Survival and weights in SMA mice treated at PND0.
Discussion
We have recently synthesized novel DNA derivatives incorporating tosyl (p-toluenesulfonyl) [53], mesyl (methanesulfonyl) [43], and busyl (1-butanesulfonyl) [45] phosphoramidate groups, respectively, and demonstrated that they can form complementary duplexes with RNA, which were more (tosyl) [53] or less (mesyl or busyl) destabilized [43,45] in comparison with the native DNA:RNA heteroduplex even if such groups replace all the phosphates in the oligonucleotide chain. In particular, when the least bulky mesyl phosphoramidate (μ) groups were incorporated into an oligodeoxynucleotide sequence, the thermal stabilities of the resulting duplexes with either DNA or RNA showed only slight differences from the corresponding native duplexes [43]. This observation prompted us to investigate mesyl phosphoramidate oligodeoxynucleotides (μ-ODN) substituted at every internucleotidic position with the μ-modification as potential RNase H-recruiting antisense agents targeting pro-oncogenic miRNAs. Our study has confirmed that μ-ODN were able to recruit RNase H-mediated miRNA cleavage with similar efficiency to phosphorothioate oligodeoxynucleotides (PS-ODNs) and better than the native DNA [42]. The μ-oligonucleotides were demonstrated to possess nuclease stability in the presence of serum, considerably exceeding that of PS-ODNs. Their antisense activity against melanoma B16 cells was also more pronounced and selective and inhibition of miR-21, miR-17, and miR-155 was longer lasting than in the case of the corresponding PS-ODNs [42]. In addition, G-rich μ-ODNs have been shown to form parallel G-quadruplexes, which were nearly as stable as the native counterparts [54].
Inspired by these results, we set out to investigate the possibility of a successful design of steric blocking antisense oligonucleotides by substituting 2′-O-methylribonucleosides (2′-OMe) or 2′-MOE ribonucleosides for 2′-deoxynucleosides in the μ-oligonucleotide backbone. Such a replacement is known to abort any RNase H activation undesirable for a splice-switching application [44]. In addition, we expected that the increase in overall hydrophobicity by introducing busyl (1-butanesulfonyl) phosphoramidate groups (β) [45] may favorably affect cellular uptake of the oligonucleotides.
Activity of ASO 2′deoxyribose and backbone modifications was measured in vitro within SMA patient-derived fibroblasts. 2′-Deoxyribose ASOs were not effective at inducing splicing modulation. This is to be expected given their RNase H recruitment activity. Both 2′-OMe and 2′-MOE ASOs actively induced splice switching of human SMN2 exon 7 when paired with the PS or μ internucleotidic linkages. Curiously, the β analog showed no activity in any ASO. Neither the endosomal escape nor intranuclear localization data could explain this observation.
It is commonly believed that ASOs enter cells through a combination of adsorptive, fluid-phase, and receptor-mediated endocytosis [55,56]. This process and intracellular trafficking are accompanied by more than 60 proteins that can bind to ASO and modulate their activity in the cell. Among them are several proteins that compete with RNase H for binding to the heteroduplex formed by PS-ASO with RNA (eg, Ku70, Ku80, P54nrb, and hnRNPs) [57], and can reduce target degradation. Also, binding to proteins affects the subcellular localization of ASO. For example, binding of paraspeckle protein P54nrb or nucleophosmin to ASO increases their accumulation in the nucleus [58,59]. On average, the increase in hydrophobicity of the 2′-substituent in ASO increased nonspecific protein binding [32]. In our study, an increase in hydrophobicity of the ASO either in 2′-deoxy →2′-OMe →2′-MOE row or from mesyl to busyl affected their cellular uptake and intracellular distribution, including nuclear localization (Figs. 3 and 4). Whereas both 2′-deoxy phosphorothioate (PS) and mesyl and busyl phosphoramidate oligonucleotides demonstrated high level of endosome release (Fig. 3A), the level of endosomal release of 2′-OMe ASOs was dependent on the backbone modification: phosphorothioate (me Nus-s) demonstrated lower endosomal release (∼60%) in comparison with ∼83% and ∼72% for mesyl (μ) and busyl (β) phosphoramidate oligonucleotides, respectively (Fig. 3B). However, in the 2′-MOE series, the μ-oligonucleotides showed the lowest endosomal release in comparison with both PS and β-series (Fig. 3C).
Analysis of the Z-stack microscopy images on the fixed cells after a 24-h incubation with Cy5-labeled ASOs demonstrated the accumulation of 2′-deoxy phosphorothioate, 2′-OMe mesyl phosphoramidate ASO, and all three variants of 2′-MOE ASO in different compartments of the cell nucleus (Fig. 4).
Differences between expected activity from cellular localization to the nucleus and measured activity may be due to the lipofectamine transfection reagent versus gymnosis uptake. Lipofectamine will aid endosomal escape, but not nuclear transport.
Subcutaneous administration of the 2′-MOE mesyl oligonucleotide isosequential to nusinersen (MOE-Nus-m) to SMA model newborn mice even at the highest evaluated dose of 40 mg/kg resulted in a median survival of only 18 days (Fig. 5A), while the 2′-MOE phosphorothioate (nusinersen) enabled median survival of the pups for 222 days (Fig. 5B). Lower doses of both ASOs (20 and 30 mg/kg) were inactive. Observed effect was sequence specific as the control scrambled and luciferase oligonucleotides did not show any activity (Fig. 5D, E). However, there were no statistically significant difference in the weights of the pups between nusinersen and its mesyl analog at 40 mg/kg (Fig. 5F).
These results could possibly be rationalized in the light of confocal microscopy data on cellular uptake and endosomal release of 2′-MOE oligonucleotides with phosphorothioate or mesyl phosphoramidate backbone in the live cells. The release level of 2′-MOE mesyl phosphoramidate oligonucleotides was ∼2 times less in comparison with 2′-MOE phosphorothioate oligonucleotides. It is suggestive of an issue with endosomal escape for this type of ASO, which may have affected their in vivo activity as potential splice-switching agents, but, of course, the cellular model we used for uptake studies is not perfect. Furthermore, the differences in biodistribution and metabolism of the phosphorothioate and mesyl phosphoramidate oligonucleotides could be responsible for the disparate in vivo results, similar to what has been shown recently by direct comparison of the in vivo efficacy of MOE PS (nusinersen) and morpholino phosphorodiamidate oligonucleotide (PMO) in a severe mouse SMA model [60]. Thus, additional studies are required to elucidate bioavailability and pharmacokinetics of mesyl phosphoramidate oligonucleotides.
The subtle differences in cytoplasmic and nuclear localization of the 2′-MOE mesyl ON compared to the 2′-MOE PS ON may reflect different patterns of protein binding and, thus, different pathways of endosomal escape and nuclear uptake for these two phosphate modifications. It may be concluded that protein binding of mesyl phosphoramidates may be less avid than for phosphorothioates and, while it may decrease systemic toxicity as it has been shown recently for RNase H-competent mesyl oligodeoxynucleotides [61], although it is not a serious concern in the case of nusinersen as it is not delivered systemically, it may also affect their antisense activity in vivo, particularly when combined with alterations in the pentose sugar from 2-deoxy to 2-MOE-ribose. As the quest for new chemistries for splice-switching applications continues to constantly produce yet untried structural combinations [62,63], it places this work, despite the somewhat discouraging in vivo results, into a wider context of elucidating “structure–activity” patterns for steric block antisense oligonucleotides with altered sugar-phosphate backbone. Thus, alternatives to the common phosphorothioates or PMOs such as mesyl and other sulfonyl phosphoramidate oligonucleotides merit further investigation as potential antisense therapeutics.
To conclude, we have found that the mesyl phosphoramidate oligo-2′-MOE ribonucleotide (2′-MOE μ-ASO) equivalent of nusinersen was not as effective as the FDA-approved 2′-MOE phosphorothioate drug in promoting long-term survival of neonatal mice in the mouse model of SMA even at the highest dose of 40 mg/kg. The low activity of 2′-MOE μ-ASO compared to nusinersen could be possibly ascribed to less efficient endosomal release and/or nuclear uptake of the 2′-O-alkyl RNA oligonucleotides having mesyl (μ) or busyl (β) phosphoramidate internucleotidic linkages as suggested by confocal microscopy study in live HEK293 cells. This may imply different pathways for intracellular trafficking and nuclear import for mesyl phosphoramidate (μ) oligonucleotides with either DNA or 2′-O-alkyl RNA backbones. The data obtained may be useful for rational design of antisense oligonucleotides with predominantly cytoplasmic or nuclear mode of action.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
S.M.H. funding comes from the Medical Research Council (MRC grant no. MR/R025312/1). The SMA mouse model and patient fibroblasts were obtained through an MTA. Microscopy studies were supported by the Institute of Cytology and Genetics (Novosibirsk, Russia) project No. 0324-2019-0042-C-01. T.S.Z. and D.A.S. acknowledge financial support from the Russian Foundation for Basic Research (D.A.S. from grant nos. 18-515-57006, 18-29-09045, and 18-29-08062, and T.S.Z. from grant no. 19-04-00298).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.