
Abstract
Trypanosoma cruzi, which causes Chagas disease, is one of the most lacerating parasites in terms of health and social impacts. New approaches for its study and treatment are urgently needed since in more than 50 years only two drugs have been approved. Genetic approaches based on antisense oligonucleotides (AONs) are promising; however, to harness their full potential the development of effective carriers is paramount. Here, we report the use of an engineered virus-like protein C-BK12 to transfect AONs into T. cruzi. Using gel electrophoresis, Dynamic Light Scattering, and atomic force microscopy, we found that C-BK12 binds AONs and forms 10–25 nm nanoparticles (NPs), which are very stable when incubated in biological media, only releasing up to 25% of AON. Fluorescence microscopy and qPCR revealed that the NPs successfully delivered AONs into epimastigotes and reduced the expression of a target gene down to 68%. Importantly, the protein did not show cytotoxicity. The combination of high stability and capability to transfect and knock down gene expression without causing cell damage and death makes the protein C-BK12 a promising starting point for the further development of safe and effective carriers to deliver AONs into T. cruzi for biological studies.
Introduction
T
Since the 1960s, the small-molecule drugs Nifurtimox and Benznidazole continue being the only drugs available for the treatment of Chagas Disease, in spite of several drawbacks such as low efficacy, numerous adverse effects, and variable effectivity against different strains of T. cruzi [4–7]. The class of drugs tested in clinical trials with limited results so far includes organic molecules [8–11]. These setbacks urge the researchers to look for more effective and safer trypanocidal drugs [12,13] by exploring different classes of molecules and alternative approaches.
Nucleic acids, such as antisense oligonucleotides (AONs) or short interfering RNA (siRNA), have brought large benefits to other fields. With several Food and Drug Administration-approved molecules, AONs are among the most advanced therapeutic alternative to treat various pathologic conditions because of their effectivity, safety, and capability to target undruggable proteins [14,15]. However, their use against T. cruzi has been largely overlooked, even though AONs are the only nucleic acid alternative available against T. cruzi, because the parasite lacks the machinery needed for processing siRNAs [16], unlike other trypanosomes such as T. brucei, where siRNAs have facilitated the functional analysis of their genes [17].
AONs are short (∼20–40 nucleotides), synthetic, single-stranded (DNA or RNA) oligonucleotides that halt the production of a target protein by inhibition of its specific mRNA translation or maturation by sequence complementarity, while leaving all other mRNA unaffected [15]. The potential of knocking down in T. cruzi the expression of specific genes with AONs has been scarcely used; there are just a few studies that report their effective use in T. cruzi for studying the role of specific genes [18–24]. However, to harness the wide potential of AONs against T. cruzi and eventually developing new and more powerful molecular genetic tools, the development of AON carriers into T. cruzi cells is paramount.
The main breakthrough needed for exploiting the inhibitory effect of AONs in T. cruzi is to effectively deliver them into the parasite cells. The most used methods to transfect AONs, or other genetic material, into T. cruzi have been diffusion through the cell membrane [18–24] or electroporation [19,25,26]. However, these methods have limited flexibility, low efficiency and cannot be translated in the future into therapeutic tools; also electroporation leads to a high degree of cell death, is time-consuming, and needs costly and specialized equipment [27]. For these reasons, they are suboptimal for therapeutic applications and even for research purposes.
Chemical methods for transfection, instead, have become an important alternative to electroporation, because they are more flexible and easier to use and can work in physiological conditions. The most advanced chemical methods for transfection are based on nanotechnology. For example, nanoparticles (NPs) based on cationic lipids or polymers (liposomes or polymersomes) have been used to internalize small-molecule drugs into T. cruzi but have not been used for nucleic acids [28]. This suggests that NPs are a valuable and promising alternative for delivering AONs into T. cruzi.
The aim of this study is to evaluate, for first time, the efficiency of an advanced recombinant protein that forms artificial virus-like particles to transfect AONs into T. cruzi. The protein used in this study, C-BK12, has the ability to mimic fundamental molecular properties of viral capsid proteins with great fidelity such as cooperative binding of single-DNA molecules and formation of highly organized and monodisperse protein-DNA nanostructures.[29–31]. C-BK12 was previously designed and consists of a highly soluble “C” collagen-like block that provides colloidal stability, and a “BK12” block, made up of 12 lysines that bind electrostatically to any nucleic acid sequence.
Delivering AONs into parasites with the help of virus-like protein-based nanocarriers may represent a novel strategy to facilitate transfection, which has been limited because of the lack of simple and flexible molecular tools. This could make the introduction of AONs of target genes for research purposes easier.
Materials and Methods
Materials
HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (Gold Biotechnology) 250 mM pH 7.4 was prepared by using deionized water. Lyophilized AONs (Integrated DNA Technologies; San Diego, CA) were dissolved with sterile MQ-water to a final concentration of 100 μM, which was used as stock solution for preparing samples. The AON 5′-GTCCTCCCTTTCCGTGCTGT-3′, which corresponds to the complementary sequence of the gen TcIP3R of T. cruzi [22], and its fluorescent version, ATTO 488 conjugated in 3′, were used in this study. The random oligonucletotide sequence was: 5′-TCCTGTGGGCCCCCTTTCTT-3′. The primers used for the qPCR were: TcIP3R-specific primers: (sense) 5′-GGAGCACTTCAGCAACATCA-3′, (antisense) 5′-GCAGCCAGTCGTAGGAGAAC-3′; GAPDH (GenBank: X52898.1):- sense: 5′- AGCATACAGGAGATCGACGC-3′, antisense: 5′-CGTAAATGGAGCTGCGGTTG-3′.
Biosynthesis and characterization of the protein
The C-BK12 protein was produced according to the previously reported methods [29,31]. Briefly, a recombinant strain of GS115 Komagataella phaffii (earlier known as Pichia pastoris) was grown in 2 L baffled flasks, and protein was produced and secreted to the extracellular media after induction with 10% v/v methanol for 72–96 h at 30°C. The supernatant of the culture was obtained by centrifugation at 10,000 g at 4°C for 20 min by using a JA-14 rotor (Beckman). The pH of the supernatant was adjusted to 8.0, and centrifugation was carried out again to precipitate salts in the medium. On filtration of the media by 0.22 μm membrane filter, the protein was purified by fractional precipitation with ammonium sulfate (60%) and acetone (40% and 80%). Each centrifugation step was performed at 30,000 g at 4°C for 20 min. Purified protein was extensively dialyzed against deionized water and then lyophilized. Usually, a yield ≈10 mg/L of cell free broth was obtained.
Molecular characterization of the protein
Integrity and purity of the purified protein was characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Briefly, approximately 20 μg of protein was loaded on to 7.5% SDS-PAGE and run at 120 V for 1.5 h. The gel was stained with SimplyBlue™ SafeStain (Invitrogen) and documented by using an Azure c300 Imaging System (CA). For MALDI-TOF, ∼, 5 μg of purified protein was analyzed in a Microflex mass spectrometer (Bruker) with a sinapinic acid matrix. The calibration was done based on Protein Calibration Standard II (Bruker).
Formation of protein-AON NPs
Aliquots of stock solutions in buffer HEPES 25 μM pH 7.4 of protein (200 μM) and AON (10 μM) were mixed in the desired protein-to-DNA charge ratio N/P, which is defined as the molar ratio of the primary ammonium ions (N) of the Lysine chains and the phosphate groups (P) in the AON. The mixture was incubated overnight (15–16 h) at room temperature (RT) before being used.
Electrophoretic mobility shift assays
The assay was run in a 15% polyacrylamide gel for 1 h at 100 V. Samples were prepared at an ATTO 488-AON concentration of 0.5 μM in 10 μL. DNA in gel was imaged by using epi-blue fluorescence using an Azure c300 Imaging System. Mobility shifts were quantified by using ImageJ software.
Dynamic light scattering of protein-AON NPs
Dynamic light scattering (DLS) measurements of samples with AON and protein concentration of 10 and 130 μM, respectively, were carried out with a Zetasizer Nano-S (Malvern Instruments, United Kingdom) equipped with a 4 mW He–Ne ion laser emitting at 633 nm. The measurements were usually the average of five runs for 30 s at 25°C taken every 5 min, using a scattering angle of 173°.
Atomic force microscopy of protein-AON NPs
Liquid samples with AON (2 μM) and AON + protein (80 μM) were deposited onto a freshly cleaved mica substrate. After 3 min of incubation, the samples were rinsed with 1 mL of Milli-Q water to remove buffer salts and non-absorbed particles; then, the substrate was dried by using compressed air stream. Samples were imaged at RT by using a MultiMode 8-HR (Bruker, MA) and silicon nitride cantilevers for ScanAsyst™ in air mode with a nominal spring constant of 0.32 Nm−1. Images and height profiles of the particles were processed by using NanoScope Analysis V.1.89. Diameter (xy dimensions) of NPs were measured with ImageJ software.
Stability of protein-AON NPs
Protein-AON NPs, with protein and AON concentrations of 80 μM and 0.5 μM, respectively, were mixed with liver infusion broth media (liver infusion tryptose; Difco) supplemented with or without 10% fetal bovine serum (FBS; Gibco) in an equal volume and incubated at RT. Sample aliquots (20 μL) were taken at specific times and stored at −20°C before being analyzed by electrophoretic mobility shift assay (EMSA). For heparin stability, aliquots (10 μL) of NPs containing AON (2 μM) and protein (80 μM) in 115 μL in HEPES 25 mM pH 7.4 were mixed with heparin aqueous solution (50 mg/mL, Inhepar®, PISA, Mexico) and filled with buffer up to 15 μL. The samples were incubated at RT for 30 min or 2 h and then analyzed by EMSA.
T. cruzi culture
Epimastigotes of Mexican strain T. cruzi TcI Qro (TBAR/MX/0000/Queretaro) were used in this study [32,33]. The parasites were cultured in LIT medium supplemented with 10% FBS and 25 μg/mL hemin and maintained for 3–4 days at 28°C to obtain the log phase of growth. To determine the parasite growth in a small volume, 107 parasites in exponential growth phase were harvested, resuspended in 20 μL of LIT medium that was not supplemented (without hemin and FBS), and incubated for 4 days at 28°C. The growth and morphology were monitored daily by determining cell density in a Neubauer chamber, and the morphology was observed in Wright-Giemsa stained parasites (Sigma-Aldrich).
Preparation of nanoparticles for studies with T. cruzi
Protein-AON NPs (N/P = 24) at the desired AON concentration (10, 20, or 40 μM) were prepared by adding the needed volume of the AON aqueous stock solution (100 μM) and buffer HEPES 25 μM pH 7.4 (and water if required) to previously weighted lyophilized protein C-BK12. Samples were mixed and homogenized by pipetting a few times, heated at 70°C for 10 min, and incubated for 19 h at RT to ensure full solubility of the protein and dispersion into monomeric subunits that are capable of assembling into NPs. For every experiment fresh NPs were always prepared.
T. cruzi transfection with NPs
For transfection assays, 107 parasites and C-BK12 + ATTO 488-AON NPs were mixed in non-supplemented LIT medium for a final volume of 20 μL and incubated for 24 h at 28°C. Parasites treated with Lipofectamine 2000 (1.5 μL) (Invitrogen), prepared according to the manufacturer's instructions, ATTO 488-AON, C-BK12 protein and buffer were used as controls. Three independent experiments were performed in duplicate. After incubation, parasites of each condition were washed twice with PBS; fixed in coated coverslips for 30 min with 4% paraformaldehyde; then washed with PBS; and finally incubated with 0.5 M glycine at RT for 1 h. DAPI (4′,6-diamidino-2-phenylindole) was used at 50 μM for 15 min at RT for staining the nucleus and kinetoplast. Slides were visualized under an Olympus IX71 epifluorescence microscope, and the images were analyzed by ImageJ software. At least 100 parasites were counted in each independent experiment to determine the percentage of parasites with normal morphology and fluorescent signal inside.
MTT reductase activity assays
Approximately 2 × 106 parasites of each treatment and controls, as mentioned earlier, were washed twice with PBS. As a positive control of nonviable cells, parasites were incubated with H2O2 at 160 μM by 15 min at 28°C. Pellets were resuspended in 25 μL of LIT medium that was not supplemented containing tetrazolium dye [MTT 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (0.83 mg/mL) and incubated for 4 h at 28°C. Parasites were recovered by centrifugation, and supernatant was discarded. Formazan salt was dissolved in 25 μL of isopropanol by shaking during 10 min at RT. Ten microliter of each sample was seeded into 96-well round-bottom plates (Costar, NY), and absorbance was measured at 595 nm with a reference of 655 nm in a microplate reader model 550, Bio-Rad [34]. For each condition, three independent experiments were performed in duplicate.
Total RNA isolation and quantitative real-time (RT-qPCR) for T. cruzi
Total RNA was isolated from 107 epimastigotes, using TRIzol (Thermo Fisher Scientific, CA), according to the manufacturer's instructions, followed by DNase I treatment. The RNA integrity was analyzed by 2% agarose gel electrophoresis in tris-acetic acid-EDTA (TAE) buffer (40 mM tris-acetate, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8), and it was visualized under UV light after ethidium bromide staining. Nucleic acid purity was assessed by the A260 nm/A280 nm ratio (acceptable >1.8). Quantitative real-time RT-PCR was performed as recently described by Arroyo-Olarte et al., [35].
DNase-treated total RNA (70 ng) was used as a template for one-step quantitative RT-PCR with a Universal KAPA SYBR FAST One-Step qRT-PCR Kit (Kapa Biosystems Boston) in a Rotor Gene 6000 (Corbett Research, Australia). The thermal program used was: 42°C for 10 min (reverse transcription) and 95°C for 3 min, followed by 45 cycles of 95°C for 5 s, 60°C for 20 s, and 72°C for 20 s. A melting curve analysis (59°C to 95°C, 0.1°C/s) was performed immediately after qRT-PCR to confirm single peak products and the absence of primer-dimers.
The random oligonucletotide was used as a negative control of the AON. In addition, the expected size of amplicons was confirmed by analysis of the real-time PCR products in 2% agarose gels in TAE and visualized under UV light after ethidium bromide staining. In all cases, GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as a reference gene [36], and target gene-expression levels were compared according to the Pfaffl method [37]. Calculations were performed by using REST software 2009, RG mode, with 10,000 permutations, and PCR efficiencies were calculated by Rotor-Gene 6000 software v.1.7.87.
Data analyses
Statistically significant differences between the means were determined by analysis of variance (ANOVA) using GraphPad Prism v. 6.01. The data were analyzed by one-way ANOVA using the Bonferroni method (P < 0.05).
Results
Formation of protein-AON complexes
We started evaluating the formation of complexes between the protein C-BK12 and a fluorescently labeled (ATTO 488) AON, which later could be used as a reporter of transfection into the interior of T. cruzi (Fig. 1). Studying the formation of complexes between the AON and the protein C-BK12 is important, because it has been previously reported only for long dsDNA molecules but not for short- and single-stranded DNA molecules [29,31].
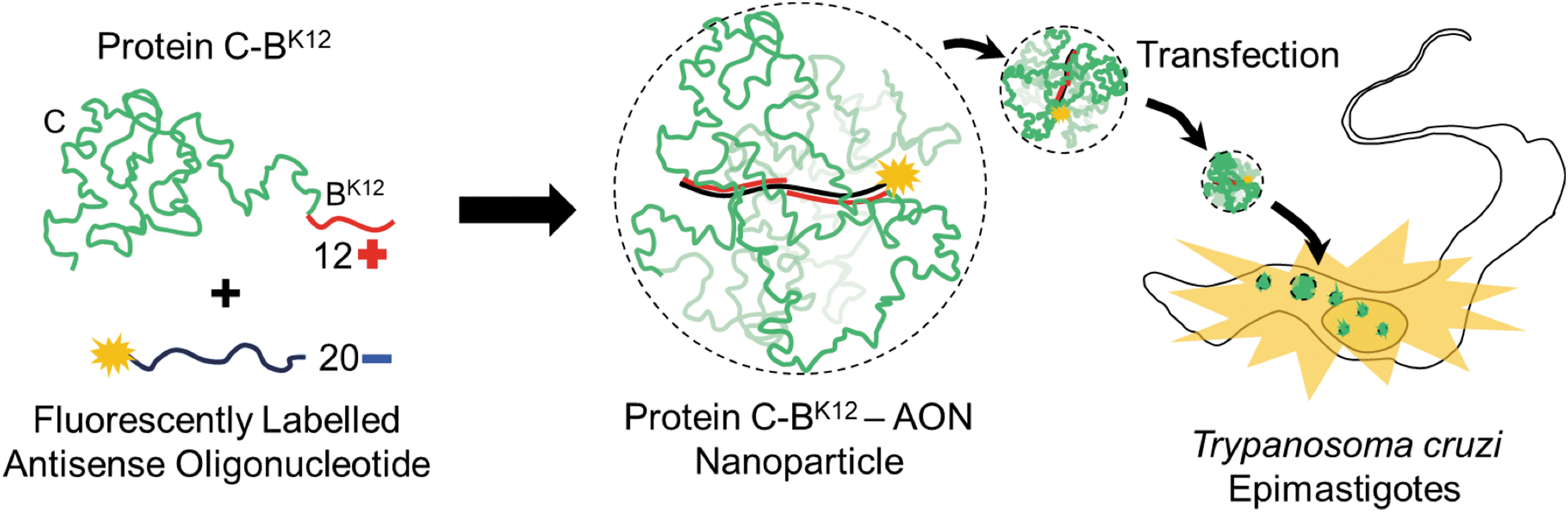
NPs based on an artificial virus-like protein C-BK12 deliver AONs into parasite Trypanosoma cruzi epimastigotes. “C” is the colloidal stability block, and “BK12” is the cationic AON binding block. AON, antisense oligonucleotide; NPs, nanoparticles. Color images are available online.
The formation of complexes between the protein C-BK12 and a fluorescently labeled AON was investigated by EMSAs (Fig. 2A). The band corresponding to the free AON disappeared with the addition of protein; and at the same time a top band, corresponding to the protein-AON complex, appeared. The latter indicated the formation of near-neutral charge complexes between the protein and the AON. An N/P > 60 (protein concentration >40 μM) was needed to completely make the band disappear and free of the AON. This represents a 60-times molar excess of cationic charge from protein over negative charge from AON. This is several fold higher than the N/P of 1–3, commonly needed to neutralize the negative charge of longer double-stranded DNA molecules [29,31,38], suggesting that the protein is weakly binding the free AON.

Protein C-BK12 binds fluorescent AONs and form near-neutral charge complexes.
For the following experiments we decided to use an N/P = 24, which has more than 85% of AON binding (Fig. 2B) as a compromise to balance possible cytotoxicity due to large protein concentrations, protein production costs, and binding most of the AON.
Protein-AON NPs
We characterized in more detail the size and shape of the formed protein-AON complexes by using molecular ensemble and single-molecule studies such as DLS and atomic force microscopy (AFM), respectively (Fig. 3 and Supplementary Fig. S1). DLS size distributions, based on number and intensity, for samples containing protein + AON complexes and protein alone revealed the presence of nano-sized particles (Fig. 3A). The occurrence of NPs was also confirmed by AFM (Fig. 3B and Supplementary Fig. S1). Although the size distributions (DLS and AFM) overlapped to some extent between protein and protein + AON samples, they had peaks of a different size.
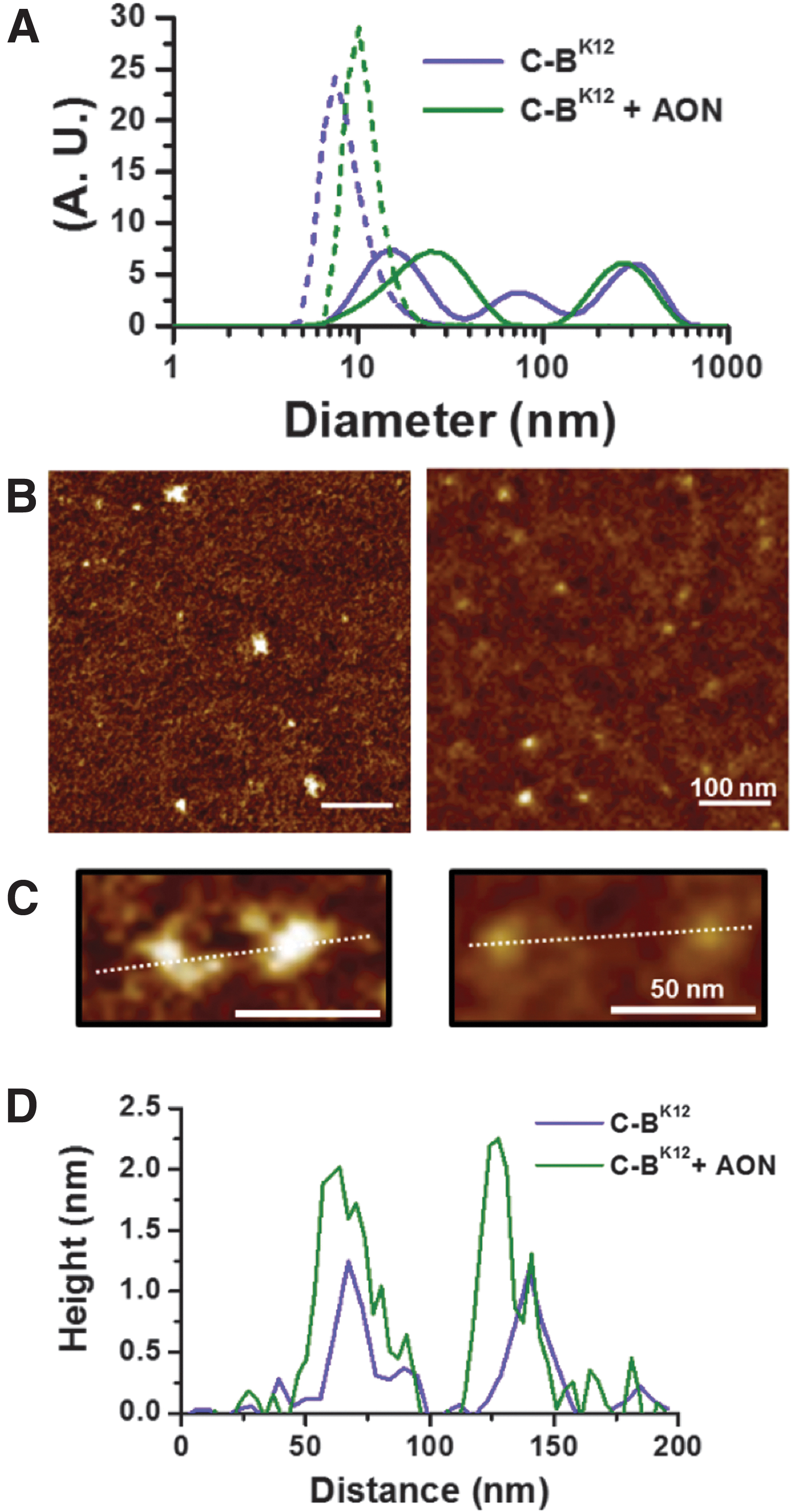
Protein C-BK12 forms NPs with AON.
The peaks observed for distributions of diameters, measured by AFM, for protein alone and protein + AON NPs were 10–20 nm and 15–20 nm, respectively (Supplementary Fig. S2A). The hydrodynamic diameter of the smaller peaks for DLS distributions based on intensity was 15.7 nm for protein and 24.7 nm for protein + AON NPs. However, for number-based DLS distributions were 7.3 nm (protein) and 10.1 nm (protein + AON NPs). It is important to point out that the smallest size peak in DLS intensity-based distributions was solely considered since the population of NPs observed by AFM indicated that the occurrence of NPs larger than 50 nm was negligible (Supplementary Fig. S2A).
According to DLS and AFM measurements, protein-AON NPs are slightly larger than protein-alone NPs. It is well known that DLS intensity-based distributions are used to give larger values and more peaks than number-based distributions, because larger particles are taken more into account than the smaller ones. The small discrepancies in the diameter values of NPs observed by AFM and DLS could be due to a change in hydrated state and flattened effects, because of their absorption on the mica wafer (AFM). In any case, regarding the results, let us assume with great confidence that the protein C-BK12 is forming together with the AON a population of NPs with a diameter between 10 and 25 nm.
Height profiles of NPs imaged by AFM (Fig. 3C, D and Supplementary Fig. S2B) show that typical protein-AON NPs are slightly higher than NPs made only of protein. When height distributions are compared for protein-AON NPs and protein alone, we noticed that they overlap (Supplementary Fig. S2B). Likewise to the measurements of diameters, protein-AON NPs coexist with protein NPs due to the large excess of protein used to neutralize the AON. Overall, the protein C-BK12 is mainly forming small-sized NPs when it binds to AON.
Stability of protein-AON NPs
To evaluate the stability of the protein-AON NPs, we followed by EMSA the release of fluorescently labeled AON from protein NPs after being incubated in LIT medium supplemented with 10% of FBS [39] (commonly used for culturing T. cruzi epimastigotes) and in LIT medium non-supplemented (Fig. 4A, B).
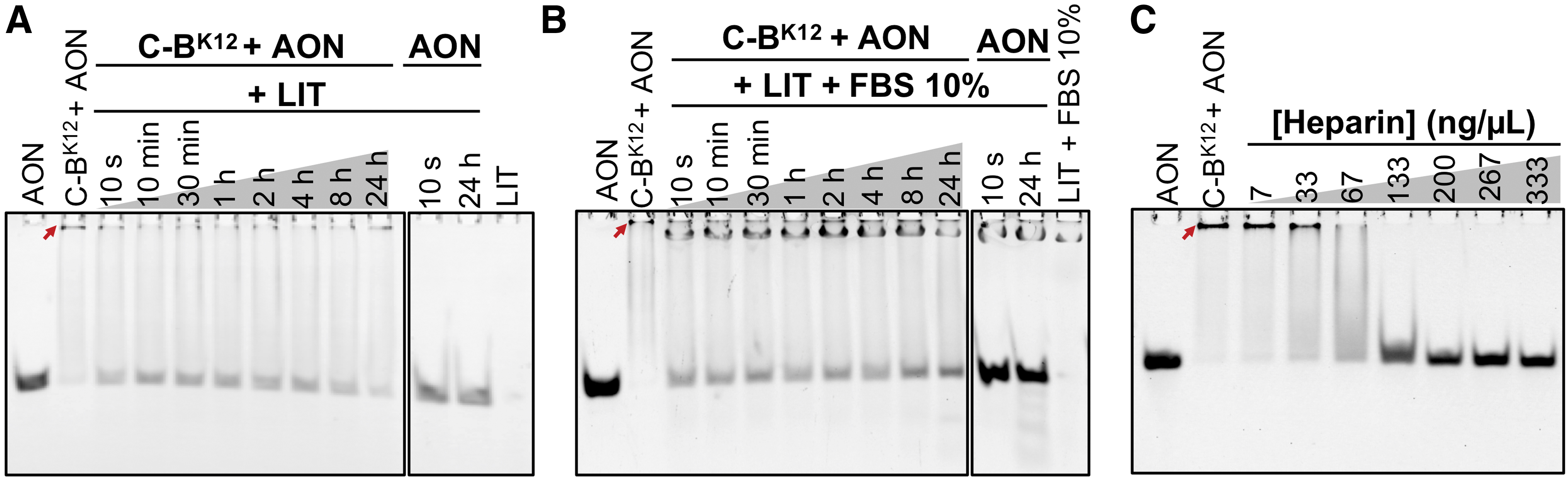
Stability of C-BK12 + AON NPs in different conditions. In
After incubation of protein-AON NPs 24 h in LIT medium, a release of up to 25% of AON was observed (Fig. 4A and Supplementary Fig. S3). We also noticed than an important proportion of the protein-AON NPs (top band) remained stable, suggesting that most of them are stable. Similarly, only up to 25% of the total AON was released from NPs incubated 24 h with LIT medium supplemented with 10% FBS (Fig. 4B and Supplementary Fig. S3). Further, protein-protected AONs showed no degradation (observed as a smeared below the free AON band) than free AON incubated in the same conditions (Supplementary Fig. S4). All these results show that most of the protein-AON NPs remain stable after incubation in media conditions and times that are relevant for in vitro T. cruzi transfection.
Heparin, a highly negatively charged polymer, is a major component of the blood that commonly destabilizes particles while they circulate in the blood stream. As heparin may act as a strong competitor with the cationic block of the protein for the AON, we tested the stability of protein-AON NPs against heparin (Fig. 4C). The EMSA revealed that we needed a concentration up to 200 μg/uL of heparin to release all the AON from the NPs (100 ng/μL for 50% disruption). This concentration is around 100–200 times higher than the concentration of heparin in blood (1–2.4 ng/μL) [40]. All these results support the idea that the protein NPs confer stability to the AON when incubated in media and times that are relevant for T. cruzi cultures.
Growth of epimastigotes in microvolumes
Before carrying out transfection tests, we standardized the culture of T. cruzi epimastigotes in volumes <1 mL. This was an important previous step since parasites are used to be grown in large volumes (>1 mL) and small volumes could cause severe stress. We confirmed that they grew normally compared with the parasites' growth in large volumes and presented usual cell motility, typical elongated morphology with terminal flagellum, and conserved organelles such as kinetoplastid before nucleus (Supplementary Fig. S5); therefore, their growth in microvolumes was viable.
Transfection of T. cruzi with NPs
Once we confirmed the normal growth in microvolumes, we evaluated the transfection capabilities of the protein-AON NPs by following the internalization into the parasites of fluorescently labeled ATTO 488-AON (Figs. 1 and 5). After cells were incubated for 24 h with various concentrations of ATTO 488-AON alone (10, 20, and 40 μM), no fluorescent signal was observed. Conversely, when we used protein-AON NPs with AON concentration of 40 μM, a green fluorescent signal was detected in 35% of parasites (Fig. 5A, B). This value is 2.3 times higher than that observed for Lipofectamine (15%), a transfective cationic lipid commonly used as a standard in mammalian cell lines (Fig. 5A, B). The fluorescent AONs internalized by the protein were observed as spots in the anterior part, and clusters at the posterior end of the T. cruzi cytoplasm (Fig. 5C).
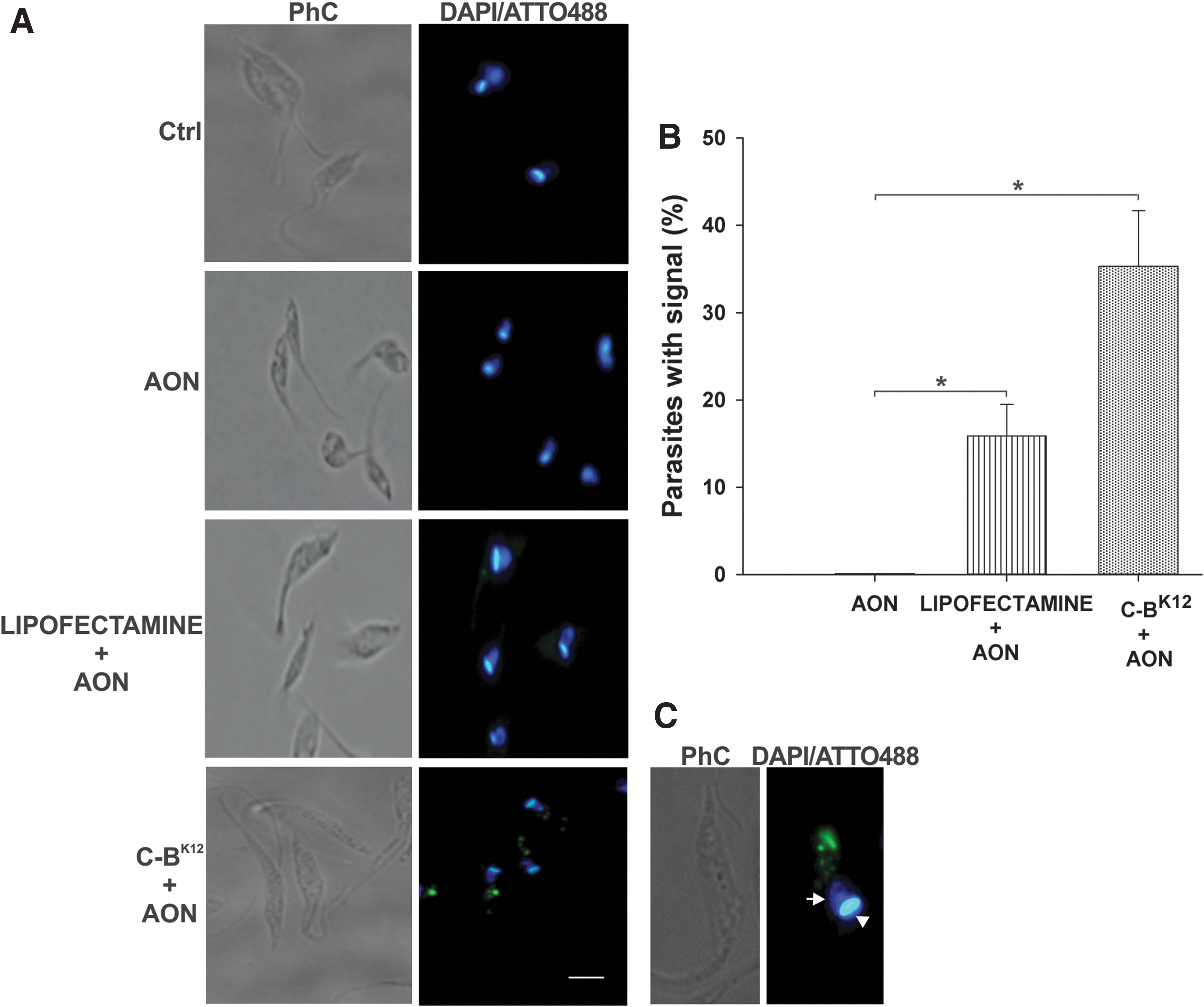
Transfection of T. cruzi epimastigotes with C-BK12 + AON NPs.
Gene knockdown by protein-AON NPs
We tested the effect of the protein-AON NPs in the mRNA expression of the target gene (Fig. 6). We chose the gen TcIP3R, because there are reported effective AONs against it [22]. Parasites transfected with the protein-AON NPs ([AON] = 40 μM and N/P = 24) reduced their mRNA expression down to 68% compared with nontreated cells. Similar mRNA reduction was observed for Lipofectamine (56%). Randomizing the AON sequence did not have an effect in the expression, confirming its specific inhibition of the target gene. Although we observed low transfection when using Lipofectamine (Fig. 5A, B), we observed a gene expression knockdown similar than our protein, but at the same time with a lower percentage of viability (Fig. 7), which is undesirable to research purposes.
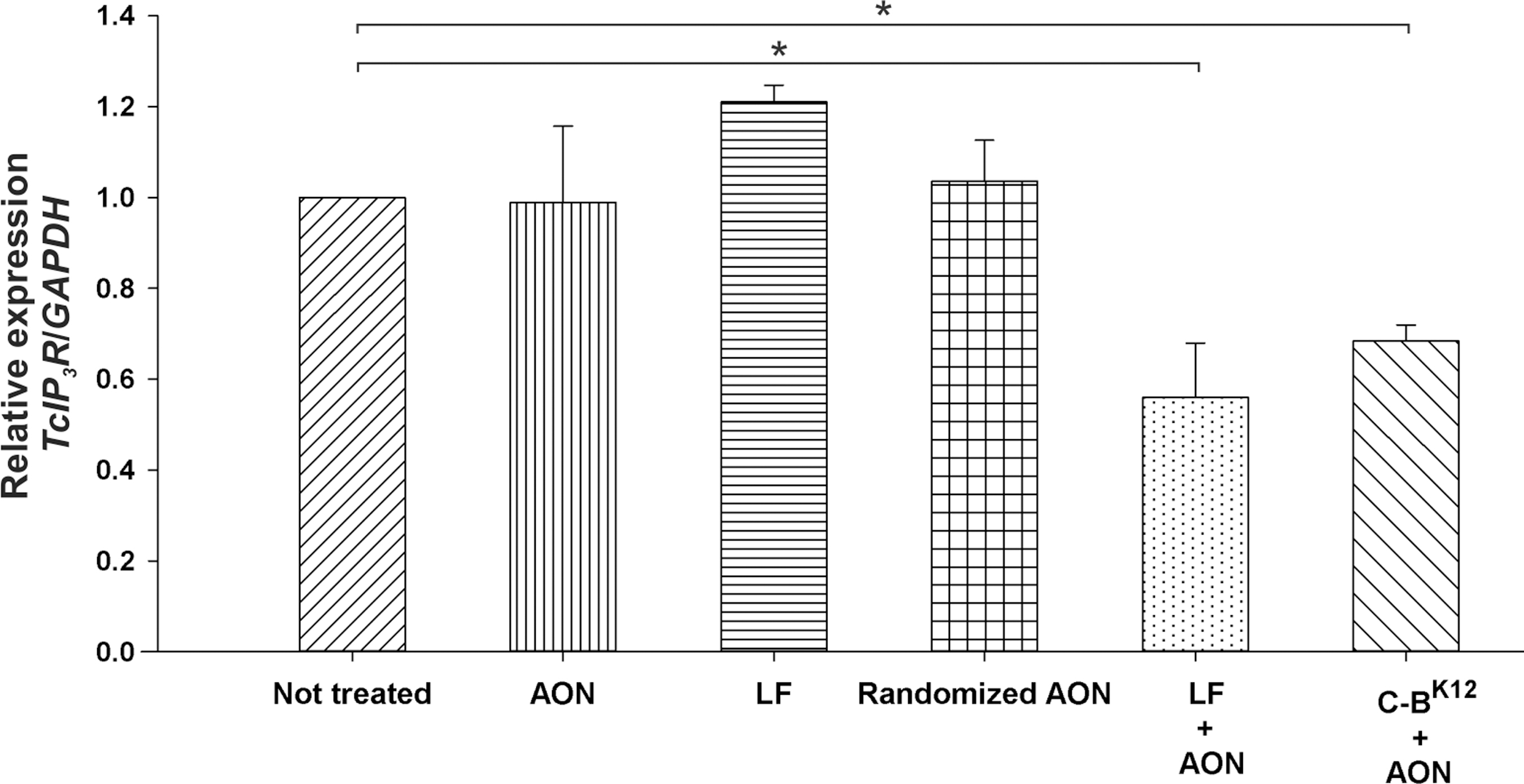
Knockdown of gene TcIPR3 expression by protein NPs loaded with AON. Quantitative determination of mRNAs was done through real-time polymerase chain reaction (qRT-PCR) after different treatments. Concentration of AON in the experiments was 40 μM. Asterisk represents statistically significant differences (P < 0.05) found between non-treated parasites and AON, Lipofectamine (LF) + AON and C-BK12 + AON NPs.
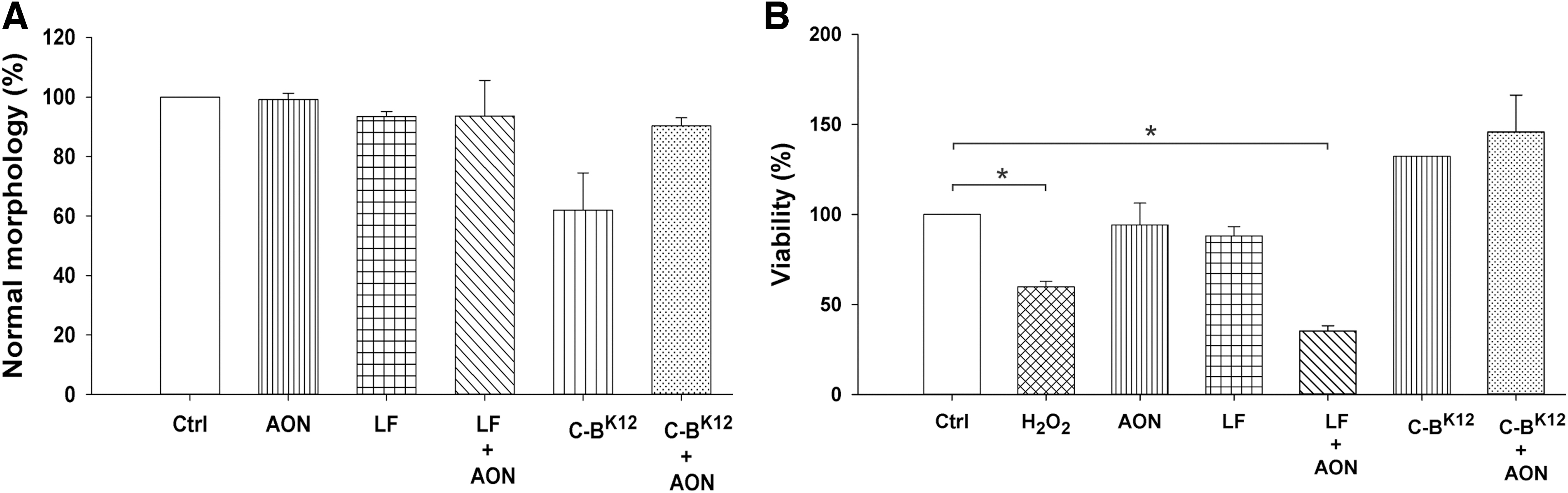
Effect of C-BK12 + AON NPs on morphology and viability of T. cruzi.
Morphology and viability of parasites after transfection
Finally, to determine whether the NPs have an undesirable toxic effect on T. cruzi after transfection, we first assessed the change from a normal elongated morphology into a round form, a typical characteristic of stressed parasites [41,42]. C-BK12 protein alone rounded 38% of parasites (a signal of stress), but this was drastically reduced to 10% for protein-AON NPs (Fig. 7). Likewise, Lipofectamine and Lipofectamine + AON only showed rounded 6% of parasites. AON alone did not induce alteration in the parasite morphology. This suggests that neutralizing the cationic charges of the carrier importantly reduces the stress caused in the parasite.
To investigate whether this morphological change was associated with reduced viability of the parasites, we performed MTT assays, which measured viability through activity of oxidoreductase enzymes. T. cruzi viability in the presence of C-BK12 protein and C-BK12 + AON NPs presented no reduction on viability (Fig. 7). When Lipofectamine alone was used, a reduction of viability of 12% was observed. In contrast, with Lipofectamine + AON, the viability was reduced to 35%; this reduction in viability is even lower than the one caused by H2O2 (60%), a known cytotoxic chemical. On the other hand, AON alone almost did not affect the parasite viability (94%). All this indicates that protein, loaded or not with AON, is completely nontoxic for T. cruzi epimastigotes, whereas Lipofectamine is toxic.
Discussion
Delivery of AONs into T. cruzi cells with protein carriers holds a large potential, because it could become an alternative tool to study the role of specific genes. However, the AON carrier needs to fulfill some requirements: (1) capability to bind the AON and form nano-sized complexes, (2) stability in relevant media or conditions for in vitro and/or in vivo experiments, (3) very low or not cytotoxicity, (4) capability to transfect the parasite, and (5) ability to release the AON in the cytoplasm or the nucleus. This work shows how the virus-like protein C-BK12 tackled, to some extent, each of the required features to deliver AONs into T. cruzi epimastigotes, as discussed later.
The artificial virus-like protein C-BK12, although it binds weakly to AONs, was able to form near-neutral charge NPs with a size between 10 and 25 nm at high N/P (Figs. 2 and 3). In spite of using high concentrations of protein to form the NPs, no signs of cytotoxicity were observed, as MTT assays and morphological inspections of protein-AON NPs-exposed parasites revealed (Fig. 7). Also, the protein exposure did not show any stress in the parasites detectable by the absence of morphological changes by optical microscopy. In contrast, the viability of the parasite was low in the presence of AON complexed with Lipofectamine. These results indicate that the transfection of parasites with C-BK12-AON NPs is viable.
Our studies showed that the protein-AON NPs are stable in medium where epimastigotes are commonly grown, such as LIT and LIT + FBS 10% (only 25% of AON was released from the protein particles) (Fig. 4). Having a high stability is very desirable, because it significantly impacts transfection efficiency. Higher stability can be achieved by using increased protein concentration or using new protein versions with stronger binding affinity for the AONs. The high stability of the protein-AON particles in the presence of heparin at blood-like concentrations and in the presence of 10% of FBS may indicate a relevant initial stability level for future in vivo studies.
Fluorescence microscopy imaging revealed that protein-AON NPs were able to enter inside the parasites (Fig. 5). The fluorescent AONs were found in the cytoplasm of the parasite, forming spots and clusters located in the anterior part and at the posterior end of T. cruzi, respectively. Previous studies using confocal microscopy in T. cruzi epimastigotes have shown that transferrin-labeled gold NPs of a similar size than our NPs (15 nm) entered T. cruzi by endocytic pathways and formed patterns similar to our protein at the bottom of the cytopharynx and the reservosomes [43,44]. This suggests that our NPs may be entering in a similar way, and that the transfection efficiency could be boosted by using ligands that enhance the cellular uptake, such as transferrin.
To compare the transfection of our protein with Lipofectamine, as the results showed, our protein-AON particles presented 2.3 times more transfection efficiency (35%) and no cytotoxicity at all after 24 h of incubation, whereas Lipofectamine had a very low transfection efficiency (15%) and high toxicity (only 35% of cell viability). The observed low transfection efficiency and high cytotoxicity of Lipofectamine in T. cruzi epimastigotes highly limits its application as transfection methodology and strikingly contrasts with the relative high transfection efficiencies achieved in mammalian cell lines. Indeed, these results are in line with the fact that Lipofectamine is not a method used to transfect T. cruzi.
Analysis of the change in expression of the target gene at the transcript level after the transfection with the protein + AON NPs reveals important details about the inhibitory gene expression effect of the AON (Fig. 6). First of all, the 32% reduction in gene expression suggests that at least some of the protein-AON NPs that are internalized into the cells at these conditions may be fully functional, meaning that the AON is released into the cytoplasm. However, we are not sure that all the internalized AON may have inhibited the target mRNA. This would depend on whether the NPs escape the endosomes efficiently, among other factors. As discussed earlier, the fact that the parasites incubated with Lipofectamine + AON had similar reduction in gene expression (36%) than our NPs (32%) may be explained by an impact caused by the high toxicity or by a quick entrance and release of the AON cargo.
The results are encouraging since this is the first report of using an engineered protein not optimized whatsoever to transfect AONs into the hard-to-transfect T. cruzi cells, which take longer to transfect with current technologies than other trypanosomes [45]. A very important aspect of this study is that it points toward different aspects of the protein that need to be optimized. Being a recombinant protein with a modular design, its performance could be largely optimized by re-engineering the current blocks or adding extra functional blocks to confer needed features. For example, single-stranded DNA-binding domains could be used for higher binding affinity for AONs; transferrin for quick, specific, and high-affinity recognition by T. cruzi receptors to achieve higher transfection capabilities [44,46,47]; and endosome escape peptides for localization of the AON in the cytoplasm.
It will be also important for analyzing these transfection conditions of the protein-AON NPs with the infective forms of trypomatigotes and amastigotes of T. cruzi. The virus-like protein C-BK12 forms stable NPs with antisense DNA, and these are able to transfect epimastigotes of the pathogenic parasite T. cruzi with apparent higher efficiency and without cytotoxicity and stress than commercially available and widely used cationic lipids. Protein-AON NPs are stable in biological media relevant for in vitro studies, and even in some in vivo related conditions and exhibited moderate inhibition of the expression of the target gene. This initial positive evaluation shows that the artificial virus-like protein C-BK12 is a promising starting point to further develop safe and effective carriers to deliver AONs into T. cruzi for experimental models to study target genes.
Footnotes
Acknowledgments
The authors thank Dr. Miguel Tapia Rodríguez of the Microscopy Facility of Instituto de Investigaciones Biomedicas (IIB) for his technical assistance in the microscopic analyses, Dr. Norma Adriana Valdez Cruz and Dr. Mauricio Alberto Trujillo Roldán of Departamento de Biologia Molecular y Biotecnologia and the Unidad de Bioprocesos (IIB) for their suggestions in the recombinant protein production, Ignacio Martinez of Departamento de Inmunologia (IIB) for technical assistance, and Laboratorio de Cromatografía de Gases y Líquidos of Instituto de Química for the MALDI-TOF analyses. The authors acknowledge the financial support for this project by CONACyT (Atención a Problemas Nacionales 2016, project 2103). B.E. acknowledges funding resources from Programa Institucional NUATEI del Instituto de Investigaciones Biomédicas, UNAM. RECG thanks UNAM-DGAPA for a post-doctoral fellowship.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Consejo Nacional de Ciencia y Tecnología (Mexico), Atención a Problemas Nacionales 2016 (2103).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.