
Abstract
A risk-based approach for routine identity testing of therapeutic oligonucleotide drug substances and drug products is described. Risk analysis of solid-phase oligonucleotide synthesis indicates that intact mass measurement is a powerful technique for confirming synthesis of the intended oligonucleotide. Further risk assessment suggests that the addition of a second, sequence-sensitive identity test, which relies on a comparison of some property of the sample to a reference standard of proven identity, results in a sufficient test of identity for most oligonucleotide drug substances and products. Alternative strategies for drug product identity testing are presented. The analysis creates a common way to communicate risk and should result in a harmonized approach to identity testing that avoids the unnecessary analytical burden associated with routine de novo sequencing, without compromising quality or patient safety.
Introduction
Synthetic oligonucleotides are an exciting new class of drugs that show promise in the treatment of cancer, cardiovascular and metabolic conditions, neurological disorders, ophthalmic diseases, and other indications. Eleven synthetic oligonucleotide drugs have reached the market* and many more are in various phases of clinical development [1].
Currently, all approved therapeutic oligonucleotides are manufactured by solid-phase synthesis. Synthesis is typically accomplished by sequential addition of nucleoside phosphoramidites [2] to a solid support derivatized with either the 3′-terminal nucleoside of the desired sequence or, more commonly, a universal linker molecule [3], which can be used to manufacture any sequence regardless of the identity of the 3′-terminal nucleoside. The synthetic steps of solid-phase synthesis are conducted using an automated oligonucleotide synthesizer, which delivers starting material (nucleoside phosphoramidites) and reagent solutions and solvents to a column reactor containing solid support. The synthesizer is controlled by a computer that executes a program specifying the quantities, flow rates, and order of addition of starting materials, reagents, and solvents to the column [4].
In size and complexity, therapeutic oligonucleotides fall between small-molecule drugs and biologics, and for this reason, regulatory guidelines that aid the development of small-molecule drugs and biologics cannot always be applied to therapeutic oligonucleotides. Recognizing the issue, the Oligonucleotide Safety Working Group (OSWG) and others have published a series of position papers providing scientific advice where current regulatory guidance is lacking. Eight position papers that focus on nonclinical issues [5–12] and two chemistry-related articles [13,14] have been published.
Recently, scientists from a group of pharmaceutical companies interested in developing therapeutic oligonucleotides established the European Pharma Oligonucleotide Consortium (EPOC). The EPOC's mission is to share knowledge and harmonize chemistry, manufacturing, and control (CMC) strategies for oligonucleotide development in hope of expediting access to life-changing medicines [15]. The consortium's initial objectives are to identify a short-list of CMC topics, convene small teams of subject matter experts to discuss current and best practices, and publish scientific advice in the form of technical white papers. An inaugural set of four CMC topics has been identified. The set includes the subject of oligonucleotide drug substance and drug product identity testing, which we define as the routine tests, analytical methods, and acceptance criteria designed to distinguish an oligonucleotide drug substance or product from other similar compounds. We report here our recommendations on the subject based on scientific rationale and our cumulative experience.
Identity Testing and Drug Substance and Drug Product Specifications
Drug substance and drug product specifications comprise a list of tests, references to analytical methods, and acceptance criteria. Regulatory advice on specifications for chemical substances is provided in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline Q6A. Q6A includes a list of universal tests applicable to all types of drug substances and drug products. Among the list is identity testing, which aims at confirming the material under consideration as the purported active ingredient.
A survey of EPOC member companies indicates that although a standard approach to routine identity testing of oligonucleotide drug substances and products is not yet established, some general trends are evident. First, the consensus opinion is that the major concern in identity testing of an oligonucleotide drug substance is confirmation of sequence. Second, there appears to be an expectation that the methods used routinely to confirm the sequence of an oligonucleotide drug substance are selective, and perhaps even specific for the intended oligonucleotide among all other oligonucleotides that might possibly be synthesized from the same starting materials. Third, and consistent with recommendations given in Q6A, there is an expectation that oligonucleotide drug substance identity testing includes a test to confirm counterion identity and quantity. In the case of drug products, expectations are less clear, but in general, there does not seem to exist the same method specificity requirement around sequence confirmation, and the use of two identification techniques, at least one of which can be defended as sensitive to oligonucleotide sequence, is usually accepted without comment. We are not aware of any requirement to confirm counterion identity or quantity for oligonucleotide drug products.
In the sections that follow, we examine the basis for the current practices regarding oligonucleotide drug substance and drug product identity testing. We enumerate likely failure modes that lead to synthesis of the incorrect sequence and describe the controls employed to prevent failure. Through the application of risk assessment, we show that de novo confirmation of drug substance sequence as part of routine batch release is overly conservative, and that more conventional approaches that rely on identity confirmation by comparison to a fully characterized reference standard provide equally effective control. We believe our proposed risk-based framework should be widely applicable to synthetic oligonucleotide drug substances and drug products and help establish a harmonized approach to identity testing in support of most marketing applications. Although the development phases are not addressed directly, an understanding of the objectives surrounding identity testing that might support marketing applications may help sponsors plan development strategies.
Risks, Failure Modes, and Controls
The application of quality risk management processes to drug substances and drug products is discussed in ICH guideline Q9: Quality Risk Management. The principles laid out in ICH Q9 form a strong, scientific foundation on which to construct a rational approach to identity testing.
An evaluation of severity, here the degree of harm to the patient associated with exposure to an incorrect oligonucleotide, is an important aspect of risk assessment. The safety and pharmacology risks associated with administering an incorrect oligonucleotide warrant the highest severity rating, and our recommendations are made with this basic assumption in mind.
For therapeutic oligonucleotide drug substances made by standard, solid-phase synthesis, there are two distinct types of risk to identity. The first risk is that associated with potential errors in the manufacturing process that lead to synthesis of an unintended sequence. The second risk concerns potential dispensing, labeling, or similar errors that result in substitution of an intended oligonucleotide drug substance for another intentionally synthesized oligonucleotide manufactured, handled, and stored in the same facility. For oligonucleotide drug products, the risks are again twofold. First, there is the risk of compounding by using an active ingredient other than the intended oligonucleotide, and second, there is the substitution risk that occurs subsequent to manufacturing, due to handling or mislabeling finished drug products.
Drug Substance
The identity risk to drug substances arising from manufacturing error can be assessed by evaluating the potential failure modes, that is, the things that can go wrong in solid-phase synthesis that lead to an incorrect sequence. The identity of an oligonucleotide made by solid-phase synthesis is determined by the identities of the starting materials and the order in which they are delivered to the column reactor. For oligonucleotides that contain both phosphorothioate diester and phosphate diester linkages, the correct order of addition of sulfurizing and oxidizing reagents is also critical. Therefore, any error that results in delivery of an incorrect starting material (or sulfurizer for oxidizer, or vice versa) constitutes a potential identity failure mode. Failure modes of solid phase oligonucleotide synthesis are shown in Fig. 1.
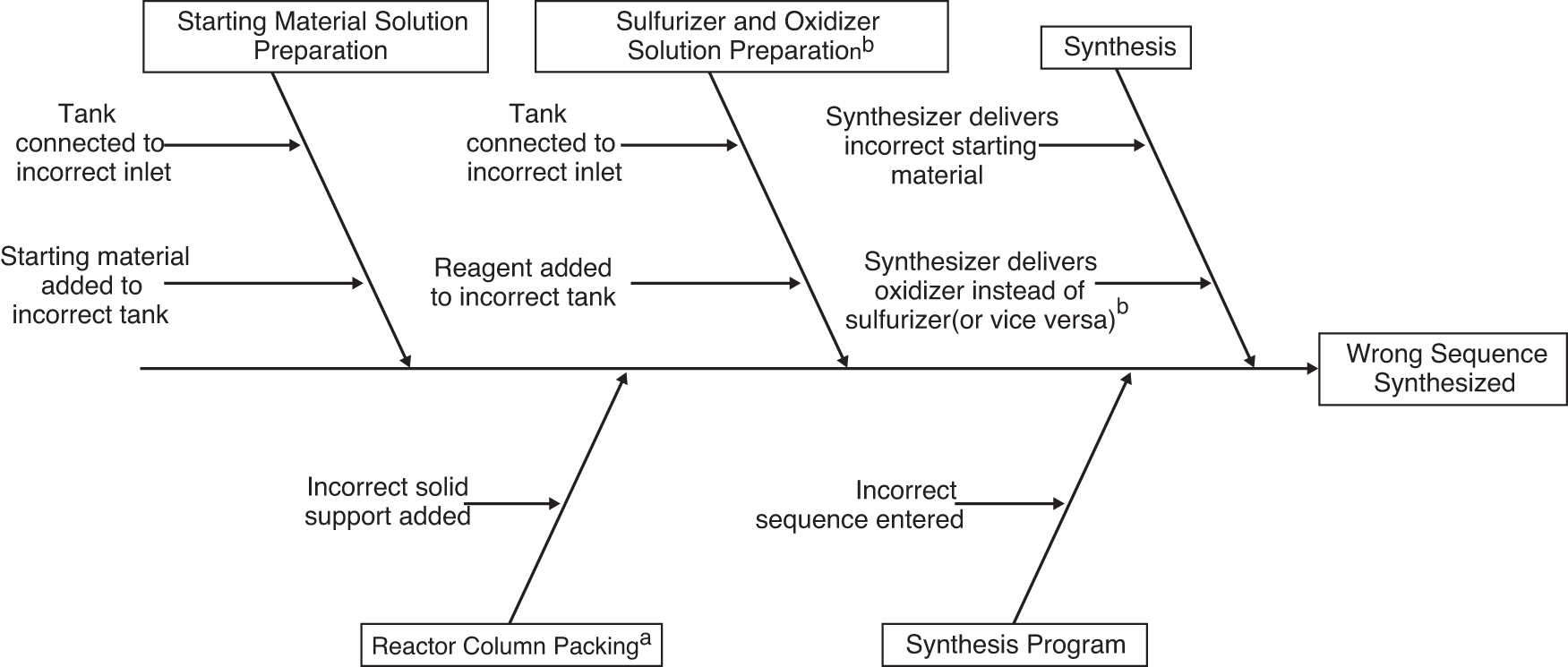
Fishbone diagram displaying failure modes of solid-phase oligonucleotide synthesis resulting in synthesis of the incorrect sequence. a Not applicable for universal solid supports. bApplicable to oligonucleotides that contain both phosphorothioate diester and phosphate diester linkages.
The next step in the risk assessment is to assign a likelihood of occurrence to each failure mode, which is best done by examining control measures designed to prevent failure. Of course, oligonucleotides destined for use in human subjects must be produced in a manner consistent with current good manufacturing practices (cGMPs), and therefore, all oligonucleotide manufacturers have in place a quality system to help ensure product quality and patient safety. Standard operating procedures (SOPs) are an integral aspect of every quality system, and cover all stages of the drug substance manufacturing process, including the operations highlighted in Fig. 1.
Starting material solution preparation is described in a manufacturing batch record, which is used to document completion of the individual steps of the process, from material receipt in the manufacturing area, through dispensing and weighing, dissolution and connection to the synthesizer. Typically, starting material solutions are prepared in stainless-steel tanks. All tanks are labeled. The calculated amounts of solvent and starting material are added to the tanks. The operation is conducted by two trained operators (4-eyes principle). After all starting material solutions are prepared, each labeled tank is connected to the corresponding labeled inlet of the synthesizer. The location of each tank is checked by two operators to ensure its connection to the correct inlet. Sulfurizer and oxidizer solutions are prepared and connected in a manner essentially identical to that used for starting material solutions. † Starting material and solution preparation steps are reviewed as a part of batch release.
The reactor column is packed with solid support. Most commonly, synthesis is conducted using a solid support derivatized with a universal linker. In such cases, the solid support does not determine the identity of the oligonucleotide synthesized, and therefore reactor column packing is not a potential failure mode. However, when a nucleoside-loaded solid support is used, the identity of the 3′-terminal residue is determined by the identity of the initially loaded nucleoside (the same is true for solid supports that contribute non-nucleoside modifiers). Consequently, addition of the incorrect nucleoside- or non-nucleoside modifier-loaded solid support to the column is a potential failure mode. Column packing is conducted by two trained operators and reviewed as a part of batch release.
The synthesis program is an electronic file of instructions required to assemble the intended oligonucleotide, including the order of addition of starting materials. Synthesis programs are created by trained operators according to SOP. The programmed order of starting material addition is verified by a second operator. Once created, synthesis programs are locked and stored in a secured location. Before synthesis, the synthesis program (now in read-only form) is uploaded to the synthesizer; second operator review ensures that the intended program has been uploaded. Confirmation that the correct program was used is conducted during batch release.
The synthesizer executes the synthesis program. The synthesizer is qualified and subject to periodic performance assessments and preventative maintenance. Synthesis progress is monitored in real time by two trained operators to ensure that all steps, including addition of starting materials, occur in the intended order. A time-log of all reagents and starting materials delivered during synthesis is compiled by the synthesizer software. The log is reviewed as a part of batch release.
A summary of failure modes and potential GMP controls designed to prevent occurrence is provided in Table 1.
Failure Modes of Solid-Phase Synthesis and Potential GMP Controls
We estimate that ∼1,600 oligonucleotides have been produced under cGMP at three large-scale oligonucleotide manufacturers over the past 15 years or so. a Our survey also indicates that no oligonucleotide of incorrect sequence was produced over the course of this manufacturing history. Therefore, there exists substantial empirical evidence of the effectiveness of the GMP controls described above. The current practice of conducting full sequencing on every batch of drug substance is inconsistent with this view, aligning instead with the notion that sequence confirmation testing should admit the possibility of any sequence, including those that can only arise by multiple failure modes. However, we believe that multiple failures are extremely unlikely in a GMP setting, and that a robust identity testing strategy can be developed by considering only a single mode of failure during any given synthesis.
We are now in a position to contemplate the probable outcome of each failure mode in terms of the incorrect sequences produced, and from this information, devise appropriate analytical testing strategies. One might initially imagine, because the sequence-specific consequences of failure depend entirely on the intended sequence, that very little generally applicable advice is possible. In most cases, however, generalizations only become difficult when one allows for more than a single failure mode to afflict the same drug substance synthesis; when one dismisses the possibility of multiple failures on the basis of the GMP controls and empirical evidence outlined above, a common theme emerges. To see how this arises, let us examine, in turn, the impact of the various failure modes.
Addition of the incorrect starting material to a starting material tank
The failure mode results in two tanks that contain the same phosphoramidite. Because a fixed number of tanks is used for each synthesis, and because multiple concurrent failures that result in swapping the contents of two or more tanks are not considered, one of the intended phosphoramidites must be omitted. Therefore, all incorrect sequences that arise by the error will have a different intact mass from that of the intended oligonucleotide.
Addition of oxidizer to sulfurizer tank (or vice versa)
The failure mode applies to the synthesis of oligonucleotides that contain both phosphorothioate diester and phosphate diester linkages, and results in two tanks that contain oxidizer and no tanks that contain sulfurizer, or two tanks that contain sulfurizer and no tanks that contain oxidizer. All incorrect sequences that arise by the error can contain only phosphate diester or phosphorothioate diester linkages, and each mis-incorporation will result in a mass difference of 16 Da relative to the intended oligonucleotide.
Incorrect nucleoside-loaded (or non-nucleoside modifier-loaded) solid support added
The failure mode results in an oligonucleotide with an incorrect 3′-terminus. Consequently, all incorrect sequences that arise by the error will have a different intact mass from that of the intended oligonucleotide.
Starting material tank connected to the wrong inlet: The outcome of a single occurrence of the failure mode is the connection of a tank to a starting material inlet not used during synthesis, which, of course, leads to synthesis failure rather than synthesis of the incorrect oligonucleotide.
‡
Swapping the positions of two tanks must be regarded as two separate instances of the same failure mode. An exception to this rule may be the rare occasion when all starting material inlets are occupied (although, in general, modern large-scale oligonucleotide synthesizers are fabricated with more starting material inlets than are required to accommodate all of the starting materials incorporated in any one sequence). In these instances, because there remains a single free inlet after the penultimate tank is connected, it may be more likely that operators swap the positions of two tanks (after all, following connection of the penultimate tank, there remains one tank and one inlet, and the remaining tank needs to be connected somewhere). If we accept that two-tank swaps represent a single instantiation of the failure mode when all starting material inlets are occupied, the possible outcomes depend on the incorporation frequencies of the starting materials. For oligonucleotides where no two starting materials are incorporated in an equal number, all two-tank swaps result in oligonucleotides of a different mass than that of the intended oligonucleotide. When two starting materials are incorporated in the same number, a single oligonucleotide isobaric with the intended oligonucleotide may arise (this occurs when the two-tank swap consists in swapping the positions of the two starting materials incorporated in an equal number). When three starting materials are incorporated in the same number, three isobaric sequences need to be considered and so on, with the potential number of isobaric sequences N increasing according to the formula
In summary, the consequences of connecting a starting material tank to the wrong starting material inlet are as follows: For oligonucleotides where the number of inlets exceeds the number of starting materials, the failure mode leads to a failed synthesis rather than an unintended sequence. When all starting material inlets are occupied, intact mass measurement may be insufficient to distinguish the intended oligonucleotide from all potential incorrect sequences; however, in the large majority of cases, only a small number of isobaric sequences need to be contemplated (see further discussion later).
Oxidizer and sulfurizer tanks connected to wrong inlet
As there is typically one inlet for each reagent, we view swapping the positions of the sulfurizer and oxidizer tanks as a single failure. Provided the intended oligonucleotide does not contain an equal number of phosphorothioate diester and phosphate diester linkages, the error leads to an oligonucleotide of different mass than that of the intended oligonucleotide. If the intended oligonucleotide does contain phosphorothioate diester and phosphate diester linkages in equal number, the control strategy needs to contemplate only one additional isobaric oligonucleotide, that is, the oligonucleotide in which the locations of all phosphorothioate diesters are replaced by phosphate diesters, and vice versa.
Synthesizer delivers incorrect starting material
Delivery of one incorrect starting material, either on a single cycle or, which is perhaps more likely, each time a particular starting material was to be delivered, must result in an oligonucleotide of different intact mass than that of the intended oligonucleotide.
Synthesizer delivers oxidizer instead of sulfurizer (or vice versa)
Incorrect delivery of oxidizer or sulfurizer on a single cycle results in an oligonucleotide with a mass 16 Da different from that of the intended oligonucleotide. Incorrect delivery of oxidizer or sulfurizer on every cycle results in an oligonucleotide with a mass difference to the intended oligonucleotide of n × 16 Da, where n is the number of phosphorothioate diester or phosphate diester linkages in the intended oligonucleotide.
Incorrect sequence entered
It is important here to consider errors associated with both initial program creation and retrieval of already created programs. The outcome of a failure during initial program creation is difficult to anticipate, although any incorrectly programmed sequence will probably be reasonably closely related to the intended oligonucleotide. The most common mistakes will likely involve inadvertent swapping of adjacent residues, or duplication and single nucleotide omission. Although the sequence-specific consequences are hard to predict, we can be certain, because the synthesis program is locked and uploaded to the synthesizer from a secure location, that the failure mode applies only to the first batch of drug substance. Therefore, full sequence confirmation of the first batch of any new drug substance is sufficient, in a GMP environment, to mitigate completely the impact of failure during initial program creation. We discuss appropriate techniques for full sequence confirmation of the first batch of a new oligonucleotide drug substance in the section on reference standard characterization (vide infra).
The consequences of uploading the synthesis program for another oligonucleotide manufactured at the same facility in place of the intended program are also difficult to predict and depend on a number of factors. In cases where the two oligonucleotides share starting materials, the error results in synthesis of an oligonucleotide manufactured at the same facility (we assume that starting materials are always connected to the same inlets). If this were to occur, the results and considerations are the same as those associated with labeling and handling errors, which are discussed in greater detail later. When the erroneously uploaded program codes for an oligonucleotide that does not share starting materials with the intended sequence, the result is not an oligonucleotide produced in the same facility, but rather one that arises from translation of the program through the intended-sequence starting materials. § The chances that the translated and intended oligonucleotides share a mass are highest when the erroneously uploaded program codes for an oligonucleotide of the same length as the intended oligonucleotide, although some overlap typically exists for sequences containing one less and one more nucleotide. In most cases, we estimate that the translated and intended oligonucleotides have less than a 1% chance of sharing a mass (Supplementary Data S1). This implies that the majority of available synthesis programs (>99%) will not lead to an isobaric impurity, and therefore, that the analytical control strategy need only contemplate one or two additional sequences.
The second risk to drug substance identity concerns handling and labeling errors that may occur subsequent to drug substance manufacturing and result in substitution of an intended oligonucleotide drug substance for another manufactured at the same facility. Many of the considerations discussed earlier in the context of mis-identity due to manufacturing errors apply to this hazard, and therefore, a similar risk-based approach is appropriate. For example, it should be possible to enumerate failure modes that might result in substitution of one oligonucleotide for another and estimate likely probabilities based on established procedural and other controls.
There is, however, one important difference, which is that unlike the set of potential oligonucleotides produced by manufacturing error, which can only be defined in probabilistic terms, the set of oligonucleotides produced at the facility is known, and therefore, the analytical challenge associated with differentiating an intended oligonucleotide from all others can be defined confidently. For example, if none of the other oligonucleotides manufactured at the facility are isobaric with the intended oligonucleotide, substitution errors can only result in replacement by an oligonucleotide of different intact mass. However, such reasoning suffers from two obvious weaknesses. First, unless the manufacturing facility is sponsor-owned or dedicated to the sponsor's products, confidentiality agreements will likely restrict the extent to which knowledge of the manufacturing portfolio can be shared. Second, even when such knowledge is available, the manufacturing portfolio will almost certainly evolve over time, so characteristics that currently differentiate the intended oligonucleotide from all others manufactured may be insufficient to do so in the future. Therefore, we recommend that all drug substance identity testing strategies include some element of future proofing to ensure the continued ability to detect potential handling and labeling errors.
The extent of future proofing required will depend on the total number of oligonucleotides manufactured; all other things being equal, the fewer oligonucleotides manufactured, the greater the likelihood that the chosen control strategy retains its specificity. Given its usefulness for detecting the consequences of failure modes associated with oligonucleotide synthesis (see Drug Substance section), it is worth considering to what extent an intact mass measurement might be considered future-proofed against handling and labeling errors. Progress in this direction requires a determination of the likelihood of manufacturing isobaric sequences in the same facility. Although it is not possible to decide the question exactly, a few simplifying assumptions permit an order of magnitude estimation. Let us assume a conservative reference class of 20-mer phosphorothioate oligonucleotides composed only of the four 2′-deoxynucleotides and their 2′-O-(2-methoxyethyl), or MOE, analogues. Let us assume, in addition, that all members must also conform to a gapmer design containing ten 2′-deoxynucleotides and 10 MOE nucleotides. We can, therefore, calculate that the reference class comprises 1.1 × 1012 possible sequences, which are distributed across 455 distinct nominal masses (Fig. 2; see also Supplementary Data S2).

Mass distribution for the reference class of 5-10-5 MOE-deoxy-MOE phosphorothioate 20-mers.
Figure 2 shows that ca. 80% of the reference class is distributed across only 120 masses (red box). Under the assumption that all oligonucleotides manufactured in the facility are enclosed by the red box, and that all oligonucleotides within the red box are equally likely to be manufactured, we can calculate the odds of two sequences sharing a nominal mass (Table 2; [16]; see also Supplementary Data S3 and Supplementary Figs. S1, S2).
Probability of Sequences Sharing a Nominal Mass
The results summarized in Table 2 show the odds that two oligonucleotides share a mass increase as the number of oligonucleotides (N) increases, reaching about 50% when N = 14. The calculations can be extended (Supplementary Data S3 and Supplementary Figs. S1, S2) to include the chances of finding higher order relations (Table 2 shows the odds up to isobaric quintets), which shows that when N = 14, there is a 2% chance of encountering an isobaric triplet. The results illustrate the following two important points. First, with any reasonable number of MOE gapmers to manufacture, isobaric pairs will occasionally arise. Second, although isobaric pairs seem almost inevitable, the odds of encountering higher order relations drop quickly, so that even with a highly constrained reference class and an extensive manufacturing portfolio, an intact mass measurement can be expected to narrow the possibilities to no more than a handful of oligonucleotides. This suggests that the combination of an intact mass measurement with another test that is reasonably sensitive to sequence should constitute a robust and future-proofed means of detecting potential handling and labeling errors.
Drug Product
The risks to drug product identity are those associated with drug substance compounding and subsequent handling and labeling of finished drug products. Compounding in error an active ingredient other than the intended oligonucleotide must result in drug product of incorrect identity. The possible outcomes and likelihood of a compounding error depend on the types and numbers of active ingredients available at the drug product manufacturing site, with the likelihood of error increasing with the number of active ingredients available (all other things being equal). We can divide the available active ingredients into oligonucleotide and non-oligonucleotide drug substances. There are several reasons to believe that the risk of compounding in error a non-oligonucleotide drug substance is negligible. As far as we are aware, all oligonucleotide drug substances are produced in facilities dedicated to the production of oligonucleotide drug substances. Consequently, all oligonucleotide drug substances available at the drug product manufacturing site are received and labeled as originating from an oligonucleotide manufacturing facility, and all non-oligonucleotide drug substances are not; the unique origin of all oligonucleotide drug substances should greatly reduce the chances of introducing in error a non-oligonucleotide drug substance to the drug product manufacturing suite.
Oligonucleotide drug product manufacturing usually proceeds by way of an initial dissolution or dilution step, which is followed by in-process measurements to confirm concentration (typically by UV absorption) and solution pH. That the physicochemical properties of any non-oligonucleotide drug substance would be sufficiently similar to those of an oligonucleotide drug substance (generally polyanionic molecules that absorb at 260 nm) to pass undetected to finished drug products seems extremely unlikely. We, therefore, propose to dismiss the risk of compounding in error non-oligonucleotide drug substances and focus on the risk posed by other oligonucleotide drug substances that might be available at the drug product manufacturing sites. Here, we are forced to acknowledge that the reasons used to dismiss the risk posed by non-oligonucleotide drug substances are reasons to inflate the risk posed by other oligonucleotide drug substances; that is, because oligonucleotide drug substances originate from a relatively small number of specialty manufacturing sites and often possess similar physicochemical properties, the chances of compounding errors are likely increased relative to small-molecule drugs (although, of course, GMP controls mean that the overall risk is extremely low).
In common with the drug substance mislabeling and mishandling example discussed earlier, the range of oligonucleotide drug substances available at the drug product site is known, and therefore, the analytical challenge of differentiating the intended drug product from all possible oligonucleotide drug products can be defined. For example, if none of the other oligonucleotide drug substances available at the drug product site are isobaric with the intended oligonucleotide, compounding errors can only result in replacement by an oligonucleotide of different intact mass. Although the strategy suffers the same weakness toward future isobaric oligonucleotide drug substances described earlier, for the reasons previously noted (Table 2), intact mass measurement can be expected to narrow the possibilities to no more than a handful of oligonucleotides. This suggests that the combination of intact mass measurement with another test reasonably sensitive to oligonucleotide sequence is a robust strategy for mitigating the risk to drug product identity due to compounding errors (see also section on Alternative Approaches To Drug Product Identity Testing).
The risk posed by labeling and handling errors of finished drug products is similar to the compounding risk, and therefore similar considerations apply. Specifically, because the physicochemical properties of non-oligonucleotide drugs are very different from oligonucleotide drugs, and because the latter often share physicochemical properties, the important challenge is to distinguish intended drug products from other oligonucleotide drug products filled at the site. Although there are perhaps means of distinguishing one drug product from another that are not applicable to drug substance, for example, vial size, fill volume, overseal color, etc., the task is essentially identical to that of detecting compounding errors, and therefore, the same approach, that is, the combination of intact mass measurement with another test that is reasonably sensitive to oligonucleotide sequence, should be sufficient.
Translating Risk Assessment Results to Analytical Control Strategies
In the previous sections, we showed how risk assessment can be used to assess the risks to oligonucleotide drug substance and drug product identity. For drug substance, the risks are those associated with failure modes of solid-phase synthesis and with subsequent handling and labeling errors. For drug products, the risks are compounding in error an oligonucleotide drug substance other than the intended oligonucleotide drug substance and mishandling and mislabeling finished drug products; therefore, drug product risks resemble drug substance handling and labeling risks.
We illustrated how an intact mass measurement is sufficient to confirm synthesis of the intended oligonucleotide drug substance, except in rare cases where a single two-tank swap, which leads to a limited number of oligonucleotides isobaric with the intended oligonucleotide, cannot be credibly dismissed. We also demonstrated that intact mass measurement is sufficient to narrow to a handful those oligonucleotides that might be substituted for the intended oligonucleotide due to mistakes in drug substance handling and labeling; drug product compounding; and finished drug product labeling and handling.
Our results suggest that it may be useful to include an intact mass measurement on the drug substance and drug product specifications. When included, the test should be able to distinguish among oligonucleotides that differ in nominal mass, which implies that mass must be measured to better than 0.5 Da. It should be acceptable to use either an absolute acceptance criterion, for example, theoretical mass ± <0.5 Da, or an acceptance criterion relative to a standard of proven identity, for example, the mass of the standard and the sample agree to <0.5 Da. The absolute acceptance criterion approach places greater demand on mass accuracy, and it will likely require use of a high-resolution mass spectrometer, whereas the relative acceptance criterion can also be supported by using a low-resolution instrument.
Our results also show that complementing intact mass measurement with a second test sensitive to oligonucleotide sequence results in a robust identity confirmation strategy (largely because intact mass measurement greatly narrows the field). We emphasize that the aim of the second identity test is to distinguish the intended oligonucleotide from a small number of potentially isobaric oligonucleotides that might be present (primarily due to labeling and handling errors in multi-product facilities). This is best done by comparing some extant property of the sample with that of a reference standard of proven identity. The strategy is closely aligned with that used for some synthetic peptides (also usually manufactured by solid-phase synthesis), where identity testing comprises intact mass measurement in combination with sample-to-standard comparison of, for example, high performance liquid chromatograph (HPLC) retention times, or mass spectrometry (MS) fragmentation patterns, or nuclear magnetic resonance (NMR) spectra. Of these, retention time is likely the least selective test for oligonucleotides, particularly among those of the same length. MS-fragmentation pattern and NMR spectral comparisons are, however, expected to provide the required selectivity.
A third technique is duplex melting temperature (Tm) analysis [17], which consists in dissolving an oligonucleotide and an equal quantity of its complementary sequence (complement) in a suitable buffer to form a double-stranded oligonucleotide (duplex). The resulting solution is heated while continually monitoring the UV absorbance. ** The absorbance of the solution increases by ∼20%–40% as the duplex “melts” into its constituent single strands. The melting temperature (Tm), which is defined as the temperature at which the sample contains equal amounts of duplex and single strands, is exquisitely sensitive to changes in base sequence, and therefore, a comparison of sample duplex and standard duplex Tm values constitutes a selective second identity test. †† Examples of the use of MS-fragmentation patterns, Tm testing, and 13C NMR to distinguish among isobaric oligonucleotides are shown in Boxes 1–3, respectively. A brief discussion of analytical method validation requirements is provided in Box 4.
While sample-standard comparison of either MS-fragmentation patterns, Tm values, or NMR spectra is likely a sufficient second identity test for most oligonucleotides, other approaches may be useful in more specialized cases. For example, oligonucleotides that contain a limited number of chiral internucleotide linkages may give rise to distinct chromatographic elution profiles or fingerprints (due to the separation of individual or groups of diastereoisomers) that could be used to help confirm identity.
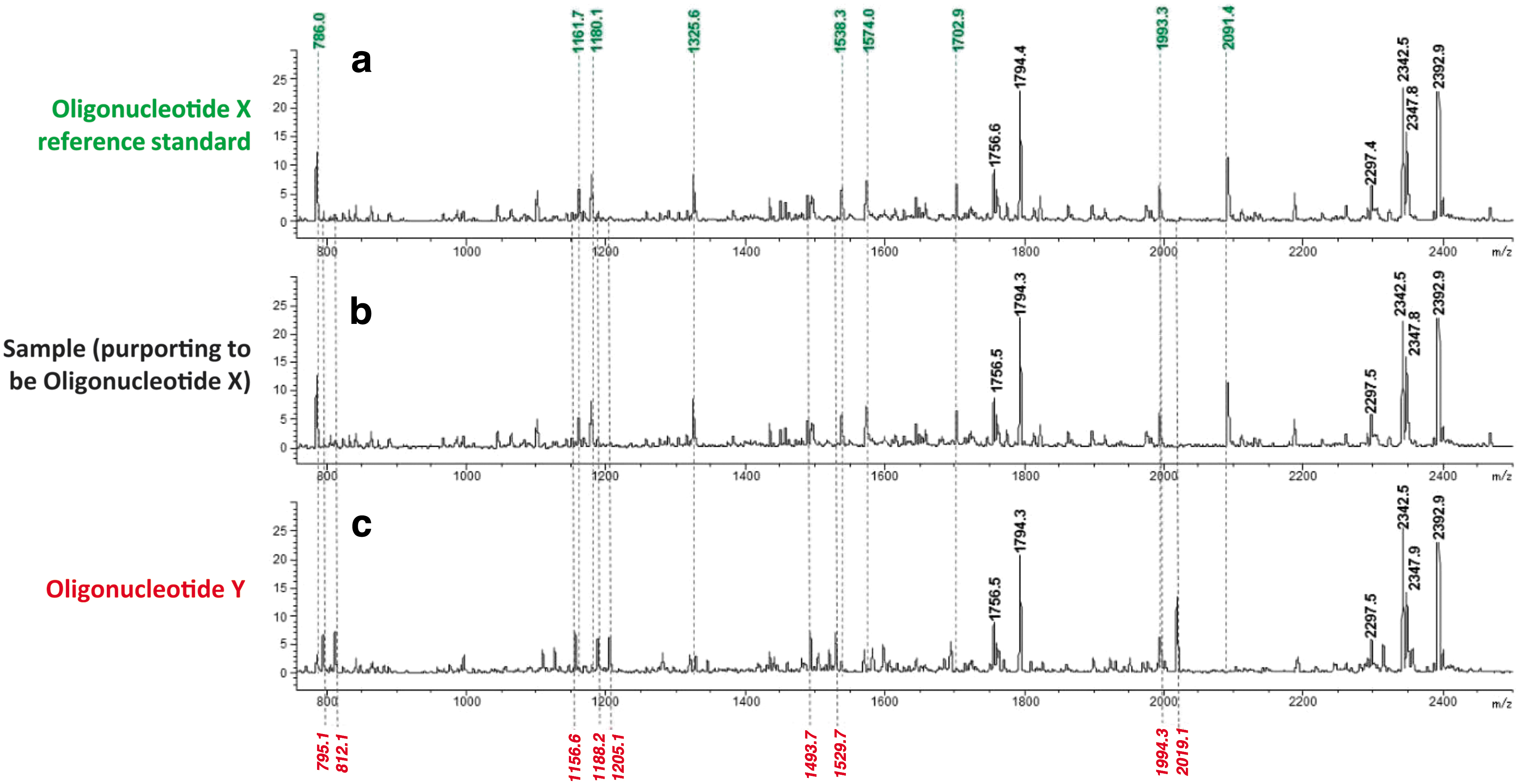
The goal of the method is to generate a mass spectrum containing a limited number of product ions that are characteristic of the intended oligonucleotide. The sample mass spectrometry (MS)-fragmentation pattern is compared with that of a reference standard of proven identity, and if the patterns match, sample identity is confirmed. The method was used to differentiate the following isobaric MOE gapmer oligonucleotides (underlined letters are 2′-O-(2-methoxyethyl)nucleotides; plain letters are 2′-deoxynucleotides; all internucleotide linkages are phosphorothioate diester):
Me Me
The MS-fragmentation pattern for the Oligonucleotide X reference standard, the sample (purporting to be Oligonucleotide X), and Oligonucleotide Y are provided in Box Figure B1 below. Fragments labeled in green and red are unique to Oligonucleotide X and Oligonucleotide Y, respectively. The data indicate that the sample MS-fragmentation pattern matches that of the Oligonucleotide X reference standard, and therefore, the identity of the sample is confirmed as Oligonucleotide X (and not Oligonucleotide Y).
Reference Standard Characterization (proof of structure)
The secondary identity tests discussed in the previous section rely on comparison of some property of the sample to that of a reference standard of proven structure. Proving the structure of a molecule as large and complex as a typical therapeutic oligonucleotide is challenging, and in all cases the proof will consist in evidence from a variety of sources. The proof should include a detailed description of the synthetic route, which begins at the point of introduction of the phosphoramidites and any other starting materials. Starting materials should be fully characterized by NMR spectroscopy (1H, 13C, 19F, and 31P, as appropriate), MS, and other relevant techniques. We recommend that the stereochemical configuration of any chiral carbon atoms created in the synthesis of starting materials be confirmed. ‡‡
Having established the identities of the starting materials, the route description should proceed to demonstrate that the proposed oligonucleotide arises as an inevitable consequence of the transformations described, according to first-principle chemical reasoning. Synthetic route evidence is especially strong for oligonucleotides, which are produced almost exclusively by an approach that has been studied in detail since its first publication more than 30 years ago [18]. The synthetic route evidence may also include process information, for example, flow rates, weights, and in-line measurements gathered during reference standard synthesis that confirm the intended quantities, flow rates, and order of addition of starting materials, reagents, and solvents to the column.
Oligonucleotides hybridize to the complementary sequence (commonly referred to as complement). A plot of UV absorbance versus increasing temperature for a solution of an oligonucleotide and its complement shows a sharp increase as the Watson-Crick duplex converts to single strands. The temperature at which the sample contains 50% duplex and 50% single strands is called the melting temperature (Tm), which is a measure of the affinity of the oligonucleotide for its complement (Box Fig. B2).
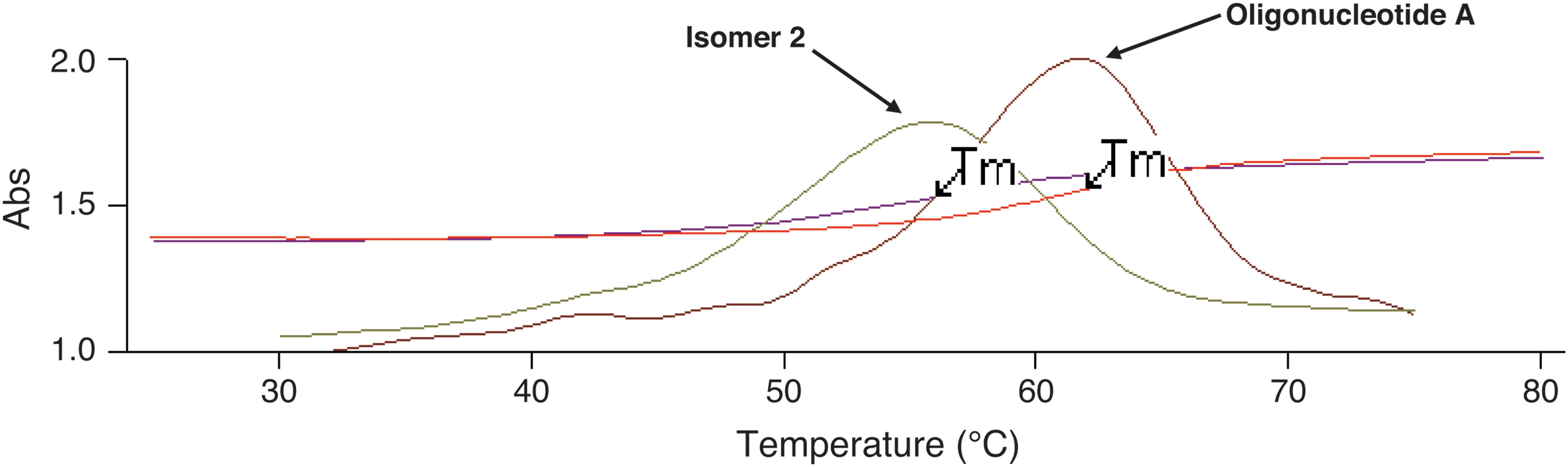
Relatively minor changes in oligonucleotide sequence significantly affect the Tm, thereby providing a basis for using Tm as an identity test. For example, the Tm value of the duplex formed by Oligonucleotide A and its DNA complement and those of the duplexes formed by Isomer 1 and Isomer 2 with the same complement are shown in Box Table B1
Tm Values of Oligonucleotide A and Isomer Duplexes
Underlined letters are 2′-O-(2-methoxyethyl)nucleotides; plain letters are 2′-deoxynucleotides; all internucleotide linkages are phosphorothioate diester; bold red letters show nucleotide swaps relative to Oligonucleotide A.
Tm value measured against phosphate diester DNA complement of Oligonucleotide A.
The results indicate that Isomer 1 and Isomer 2, which cannot be distinguished from Oligonucleotide A by mass measurement, are nevertheless easily differentiated by Tm testing.
Proof of structure will, of course, also rest heavily on analytical evidence obtained from a number of techniques. The evidence can include the results of a high-resolution mass measurement, which should be consistent with the expected molecular formula. Proton (1H) and carbon (13C) NMR spectroscopy results also contribute meaningfully to all structural claims. Given the size and complexity of most oligonucleotides, we recommend the use of a relatively high-field instrument, for example, 500 MHz (for 1H). Selected two-dimensional NMR experiments such as correlation spectroscopy, heteronuclear multiple quantum coherence, and heteronuclear multiple-bond correlation spectroscopy can be used to guide interpretation of the 1H and 13C spectra and to confirm resonance assignments. Phosphorus (31P) NMR is useful for establishing the chemical nature of internucleotide linkages, with common chemistries such as phosphorothioate diester, phosphate diester, and phosphorodiamidate exhibiting characteristic, easily quantifiable resonances. For oligonucleotides that contain more than one type of internucleotide linkage, integration of the 31P NMR spectrum provides evidence of their intended ratio. Fluorine (19F) NMR may also be useful in some instances. Finally, the nucleotide sequence of the reference standard should be confirmed by some suitable technique. A common approach is tandem mass spectrometry (MS/MS) sequencing (for an excellent review see [19]), although failure sequence analysis [20] and in-line monitoring of reactor column input or output during solid phase synthesis [21–23] are viable alternatives (see Alternative Approaches to Drug Substance Identity Testing).
Counterion Testing
With the exception of eteplirsen and golodirsen, which are neutral phosphorodiamidate morpholino oligonucleotides, all other approved therapeutic oligonucleotides are composed of nucleosides linked via phosphate diester or phosphorothioate diester internucleotide linkages. The pKa of an isolated phosphodiester is <2 [24], and all evidence is that synthetic oligonucleotides isolated from solution at or near neutral pH contain one counterion per internucleotide linkage.
Owing to its excellent sensitivity to chemical structure, NMR spectroscopy can be applied to the identity testing of oligonucleotides. With appropriate method development to optimize temperature, pH and sample concentration sufficiently resolved NMR spectra can be obtained to distinguish even closely related oligonucleotides. Box Figure B3a shows the spectra for a parent compound and 3 switch sequences [letters in square brackets are constrained ethyl nucleotides; plain letters are 2′-deoxynucleotides; all internucleotide linkages are phosphorothioate diester].
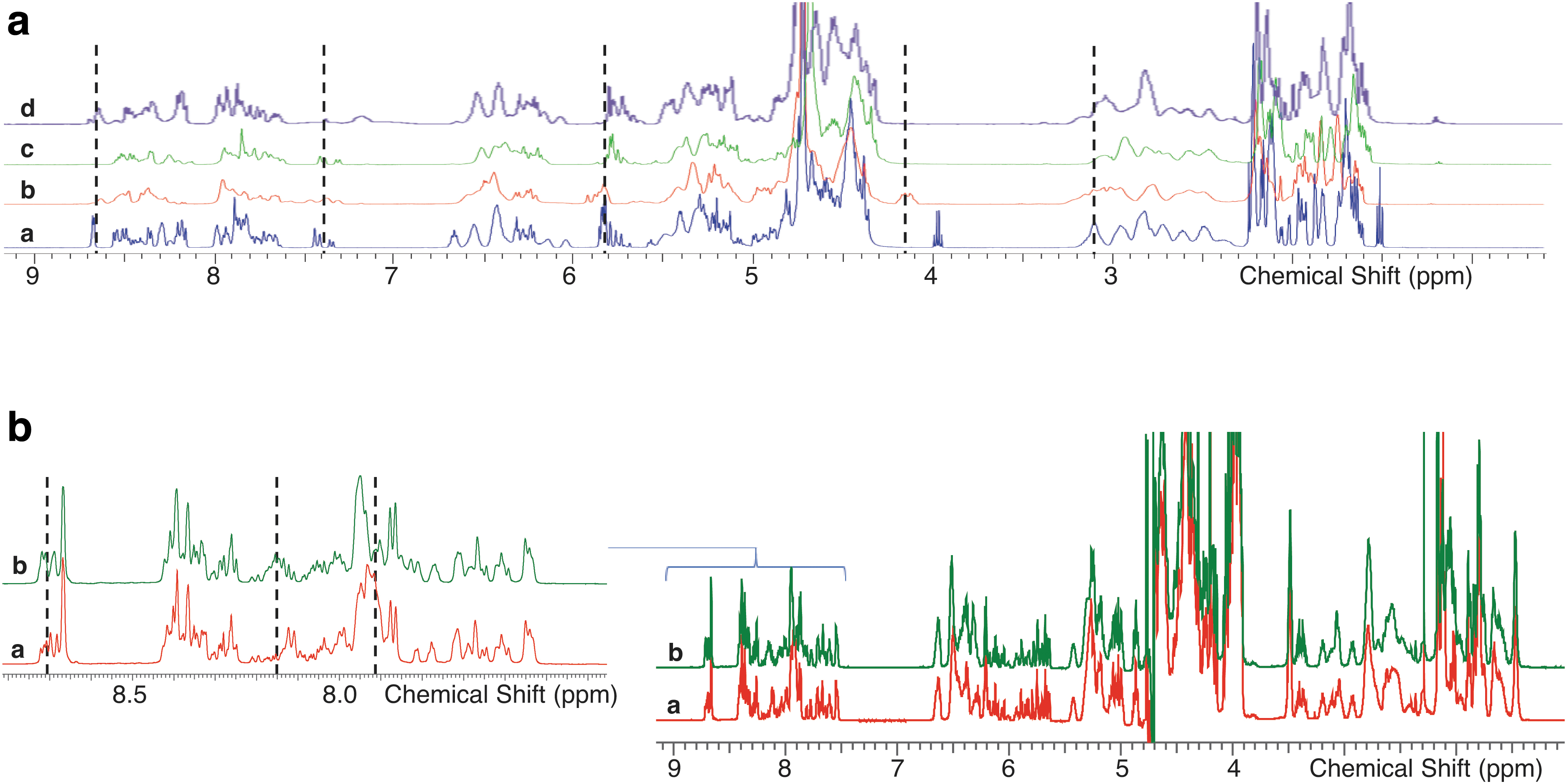
Distinct differences are observed in the resonances associated with the aromatic base protons (∼7–9 ppm), the sugar protons (∼2.5–7 ppm), and the methyl protons of the MeC and T/MeU bases (∼1.5–2.5 ppm). These regions offer a unique fingerprint of each sequence, allowing positive discrimination between oligonucleotides and confirmation of identity of a target oligonucleotide when compared with a reference spectrum.
Spectral differences are still apparent for conjugated oligonucleotides (Box Figure B3b) and where the overall structural change is small [Box Figure B3b, letters in curly brackets are locked nucleic acid nucleotides; plain letters are 2′-deoxynucleotides; all internucleotide linkages are phosphorothioate or phosphate diester].
The examples shown represent challenging tests of specificity, likely exceeding that required for routine use in a manufacturing setting, and demonstrate the power of NMR to detect subtle changes in sequence. This sensitivity to structure allows confirmation of identity of a target sequence when compared with a fully characterized reference material.
For small-molecule drug substances isolated in their salt form, a test of identification of counterion is customary included as a part of release testing. The inclusion of a quantitative test for counterion is not usually necessary, presumably because most small-molecule drug substances isolated as their salts are only singly, or occasionally, doubly charged. In such cases, weight-based drug substance assay measurements are often sufficiently sensitive to detect changes in the amount of counterion present, and therefore, an identification test that is specific for the salt itself is sufficient. The situation is more complicated for polyanionic molecules such as oligonucleotides, where it is difficult to rely on a qualitative identity test and the drug substance assay measurement to confirm counterion composition. Therefore, for oligonucleotides isolated in their salt form, we suggest that a quantitative test for counterion be included in the drug substance specification. Commonly used methods for quantitative counterion testing of oligonucleotide drug substances include inductively optical emission spectroscopy and atomic absorbance spectroscopy [25]. Quantitative 23Na NMR can also be used. Sodium counterion testing should not be required for solution drug substances.
Alternative Approaches To Drug Substance Identity Testing
Our intention in this article is to describe an approach to routine identity testing of oligonucleotide drug substances and drug products based on a modern risk-based rationale. A main objective is to separate routine release testing, that is, batch-to-batch confirmation of identity, from formal and comprehensive proof-of-structure studies conducted to establish unequivocally the structure of a reference standard. We hope to convince the reader that routine drug substance identity testing can be accomplished by combining intact mass measurement with a second test, sensitive to sequence, wherein some property of the sample, for example, MS-fragmentation pattern, is compared with that of a standard of proven identity.
We also discuss counterion identity testing, which is a required element of solid drug substance release testing of negatively charged oligonucleotides. Here, we show that the polyanionic nature of oligonucleotides requires use of a quantitative counterion test (rather than a simple measurement of identity). To the best of our knowledge, quantitative counterion testing is included at release for all approved, negatively charged therapeutic oligonucleotide solid drug substances, and therefore, our recommendations are aligned with current practice. This is in contrast to our suggestions on sequence confirmation, where drug substance specifications for all approved therapeutic oligonucleotides include some type of de novo sequence test. We have discussed at length that we regard the current, batch-to-batch practice of de novo sequencing unnecessary; nevertheless, for the sake of completeness and to avoid creating the impression that we view the current testing paradigm as inadequate, we discuss here briefly methods sometimes used for routine de novo sequencing.
The most common de novo sequencing technique is MS/MS sequencing. Although a variety of procedures have been employed, all rely on the creation of fragments of the parent (precursor) oligonucleotide, which are sorted and identified using MS. Early methodologies utilized matrix-assisted laser desorption ionization time-of-flight-MS to analyze fragments produced by partial enzymatic digestion [26]. §§ This and similar chemical degradation/MS-based techniques [27] have largely been supplanted by MS/MS, where fragments are produced in the gas phase. In the first stage of a typical MS/MS analysis, the mass spectrometer is used to isolate ions of the parent molecule by mass-to-charge ratio (m/z). Precursor ions are transferred to a collision cell, where they are fragmented by colliding with gas atoms or other molecules or by laser light or electron beam. Oligonucleotide fragmentation proceeds according to well-established pathways to give product ion fragments that are assessed by the mass analyzer. The resulting spectra are examined for the expected mass signals, and, if the expected signals are observed, the sequence of the oligonucleotide is confirmed.
An alternative means of establishing sequence is failure sequence analysis, which was first suggested in 1996 by Martin [20]. The analysis relies on the observation that step-wise coupling yields in oligonucleotide synthesis are not quantitative and, consequently, the crude products of synthesis contain small quantities (on the order of 1–2 mole% each) of oligonucleotides that failed to extend during each coupling reaction. The mass difference between failure sequences formed in adjacent cycles corresponds to the mass of the nucleotide added in that cycle, and therefore, mass differences between consecutive failure sequences are a faithful history of the order of nucleotide addition. *** Failure sequence analysis is commonly conducted using ion pair HPLC electrospray ionization mass spectrometry.
In addition to the off-line techniques described above, there have been several attempts to confirm sequence in real time, by monitoring reactor column input or output during solid-phase synthesis to confirm that the correct starting materials and reagents are delivered in the correct order. Early efforts toward this end relied on MS [21]. More recently, a group at GSK was able to differentiate among adenine, guanine, cytosine, and uracil 2′-OCH3 phosphoramidites in real time, using on-line mid-infrared (IR) and Raman spectroscopy to construct multivariate statistical process control models [22]. A similar approach was described by Stolee, who demonstrated discrimination among eight different phosphoramidites using mid-IR [23].
Because we believe release testing of drug substance need not include de novo sequencing, we regard routine inclusion of MS/MS sequencing, failure sequence analysis, or in-line monitoring as overkill. By taking this position we, of course, do not wish to indicate that such methods should not or cannot be used, only that they are unnecessary for drug substance synthesized and handled under cGMP. When used, we believe MS/MS sequencing of drug substance should be regarded as a sufficient means of confirming identity, that is, when MS/MS sequencing is conducted directly on the drug substance, no additional drug substance identity tests should be required. Failure sequence analysis and in-line monitoring of reactor input or output can readily be demonstrated specific for the intended oligonucleotide, and therefore, either method should be an acceptable substitute for direct MS/MS sequencing of drug substances. ††† However, because the methods are applied upstream of the final drug substance, the use of failure sequence analysis or in-line monitoring necessitates additional release testing of the drug substance or product (to mitigate against potential handling and labeling errors) using a method known to be highly sequence selective, such as Tm or MS-fragmentation pattern analysis.
The defining validation characteristic of an analytical procedure designed to confirm identity is specificity, which is the ability to discriminate the intended compound from closely related structures that are likely to be present. It is important to remember that a lack of specificity observed with one method may be compensated by the application of one or more additional methods, provided that the combination is specific. In the case of therapeutic oligonucleotides, closely related structures likely to be present comprise oligonucleotides arising from (a) potential errors in the manufacturing process, and (b) potential dispensing, labeling, or similar errors that result in substitution of the intended oligonucleotide for another manufactured, handled, and stored in the same facility.
We show that a simple intact mass measurement discriminates the intended oligonucleotide from most unintended oligonucleotides that are likely to be present. Here, analytical method validation should consist of a demonstration of mass accuracy that is sufficient to support an acceptance criterion (specification limit) commensurate with ruling out non-isobaric oligonucleotides. The acceptance criterion could take the form of an absolute value, or more likely, a value measured relative to a reference standard of confirmed identity, but in either case, method validation can be conducted by using the intended oligonucleotide alone. For example, if one wished to validate the ability to distinguish the intended oligonucleotide from those that differ in nominal mass, one would need to validate the ability to measure the mass of the intended oligonucleotide (or the mass difference relative to a reference standard) to better than 0.5 Da.
Intact mass measurement cannot distinguish the intended oligonucleotide from isobaric oligonucleotides likely to be present, and therefore, we recommend the addition of a second identity test recognized to be sensitive to changes in sequence. Although the extent to which it makes sense to conduct an empirical validation of specificity for the second identity test will vary among oligonucleotides and manufacturing scenarios, we offer the following general advice regarding the choice of challenge sequences, that is, sequences for which specificity should be evaluated experimentally:
All isobaric sequences arising from a single failure mode of the manufacturing process should be included as challenge sequences. For example, in cases where swapping the locations of two starting material tanks constitutes a single failure (this occurs when all starting material inlets are occupied), all All isobaric oligonucleotides manufactured by (or for) the sponsor at the manufacturing facility should be included as challenge sequences. Given confidentiality agreements, it will generally not be practical to include as challenge sequences isobaric oligonucleotides made at the facility for other sponsors.
Consideration of the first two points will often reveal no isobaric sequences. In these instances, the sponsor should document the absence of isobaric sequences as justification for omitting a formal assessment of specificity. In all cases, and to prevent erroneous rejection of the intended oligonucleotide, validation of the second identity test should also include an assessment of the likelihood of generating false negatives.
Alternative Approaches To Drug Product Identity Testing
We have discussed at length the usefulness of intact mass measurement as a part of drug product identity testing. However, unlike drug substance identity testing, drug product identity testing does not have to mitigate the risk associated with synthesis of an incorrect sequence (rather the task is to differentiate among intended oligonucleotides handled at the same facility). Because the utility of intact mass measurement is primarily to mitigate synthesis errors, we believe that non-MS approaches to drug product identity testing may be included as viable alternatives. Factors such as the ability of a sponsor to maintain full visibility to other oligonucleotide drug substances and drug products handled and manufactured at the drug product filling site should be considered. In instances where internal drug product manufacturing resources are employed, for example, the sponsor will presumably possess full knowledge of the manufacturing portfolio. Access to such information may facilitate implementation of analytical combinations that might otherwise be regarded as insufficiently selective. For example, access to other oligonucleotides handled on site may permit the development of chromatographic methods that are capable of differentiating the intended oligonucleotide from the rest of the portfolio. Substitution of intact mass measurement for other techniques may, for example, facilitate in-country identity testing in regions where MS is not readily available.
Special Considerations for Double-Stranded Oligonucleotides
Double-stranded oligonucleotide therapeutics consist of two complementary single-stranded oligonucleotides. Each strand is synthesized independently, and the two strands are annealed to form the duplex, which is taken as the drug substance. The risks to drug substance identity are those associated with synthesis, including the annealing step, and with subsequent substitution of one drug substance for another. The risks to drug product identity comprise compounding errors and substitution of one drug product for another.
Because the individual single strands of a double-stranded oligonucleotide drug substance are synthesized by the processes used to make single-stranded oligonucleotide drug substances, the failure modes and controls outlined earlier apply directly. Consequently, intact mass measurement of the individual single strands before annealing is sufficient in most cases, to confirm the synthesis of the correct sequences, and in all cases, to narrow the range to a small number of isobaric alternatives. The subsequent annealing step of the two single strands is, in and of itself, a second, independent confirmation of the sequence identities of the individual single strands, as the odds that combining anything other than the two intended single strands should result in a stable duplex seem too remote to consider. Therefore, intact mass measurement of the individual single strands combined with Tm analysis of the resulting double-stranded drug substance should be sufficient to confirm the identities of the individual single strands.
The annealing of two single-stranded oligonucleotides clearly cannot alter their identities, and therefore, the only additional failure mode associated with synthesis of double-stranded drug substance is to combine incorrect single strands. However, as discussed earlier, combining anything other than the two intended single strands will almost certainly not result in a stable duplex; therefore, Tm analysis of the duplex, in addition to confirming synthesis of the correct single strands, is a powerful method for confirming that the intended single strands were used in the final annealing step of drug substance synthesis.
The task of distinguishing among intended double-stranded drug substances resembles that described earlier for single-stranded drug substances. Intact mass measurement, which is powerful means of narrowing the range of potential single-stranded drug substances, is even more powerful for double-stranded drug substances, where the potential to measure not only the mass of the intact duplex but also those of the individual single strands exists. Nevertheless, the possibility for isobaric duplexes composed of isobaric single strands remains and, therefore, sole reliance on intact mass measurement is likely insufficient (and almost certainly insufficiently future-proofed). Therefore, orthogonal techniques, such as denaturing HPLC or Tm, are also recommended.
In most cases, Tm analysis will be also conducted, and such analysis will likely further refine the range of (isobaric) possibilities. There is, however, a subtle but important difference between the use of Tm for identity testing of single- and double-stranded drug substances. In the former, testing is conducted by addition of a characterized complement of the intended sequence, and therefore, a perfect duplex forms only in the event that the sample has the correct identity. In contrast, in the case of a double-stranded drug substance, a perfect duplex is present regardless of sample identity (because the sample is a duplex). As it is not uncommon for duplexes to have similar Tm values, the selectivity of Tm testing among double-stranded drug substances is diminished relative to single-stranded drug substances. By highlighting this difference in the application of Tm testing, the authors do not wish to suggest that the combination of Tm and intact mass measure is generally insufficient for double-stranded drug substance, but only rather that additional careful consideration is required.
Special Considerations for Long Single Guide RNA
Single-guide ribonucleic acids (sgRNAs), which are ∼100 to 110 nucleotides long, are used in CRISPR/Cas9 genome engineering technology [28]. The sgRNA directs the Cas9 endonuclease to specific endogenous genomic loci to create double-strand breaks, acting similar to a pair of molecular scissors to insert, remove, or replace specific pieces of DNA. No CRISPR/Cas9-based therapy is yet commercially approved. However, because the field is evolving and progressing rapidly, and because sgRNA are produced by solid-phase oligonucleotide synthesis, a few preliminary and cautious words regarding identity testing seem appropriate. Because of the relative length of sgRNA, sequence confirmation with sufficient coverage by tandem MS/MS is currently impractical. Next Generation Sequencing (NGS) analysis methods have, however, been adapted and applied. Although the technology and data processing require some expertise to implement, the read-out is a powerful and unambiguous confirmation of sequence, at least for unmodified RNA. ‡‡‡ Other potential techniques include MS analysis of fragments generated by partial enzymatic or chemical digestion and real-time monitoring of reactor column input or output.
Conclusions
A major goal of chemical drug development is to ensure that every dose of drug product contains a safe and efficacious quantity of the intended therapeutic. This is achieved, in large part, by building quality into manufacturing processes from the beginning, through the accumulation of scientific understanding and implementation of attribute, procedural, and parametric controls. However, even well-understood processes and carefully designed control strategies can fail, and therefore drug substances and drug products are subject to certain release tests. Release testing is primarily confirmatory in nature; that is, the main goal is to demonstrate that manufacturing processes and controls proceeded and functioned as intended.
We address here release testing requirements for identity testing of therapeutic oligonucleotide drug substances and drug products. Current practices around identity testing of oligonucleotide drug substances include what amounts to batch-wise de novo sequence confirmation, which to date, at least for approved therapeutics, has been accomplished by direct MS/MS sequencing or failure sequence analysis. In our view, de novo sequencing is beyond what would usually be regarded as confirmatory testing, and instead is consistent with the types of analyses undertaken as a part of proof-of-structure or characterization studies. The practice of de novo sequencing of oligonucleotide drug substances is also beyond standard practices employed for routine identity testing of peptide drug substances, which are also manufactured by solid-phase synthesis.
To understand and potentially redress the different practices applied to oligonucleotide and peptide drug substances, we conducted a risk assessment of solid-phase oligonucleotide manufacturing and the subsequent handling and labeling operations. For single-stranded oligonucleotides, our assessment shows that intact mass measurement can be coupled with a second technique, wherein some sequence-sensitive property of the sample, for example, MS-fragmentation pattern, Tm or NMR spectrum, is compared with that of a fully characterized reference standard to provide a sufficient test of identity. Similar reasoning suggests that the same analytical approach can be used for routine identity testing of single-stranded oligonucleotide drug products, although the simplified task of drug product identity testing relative to drug substance identity testing indicates that strategies that forgo intact mass measurement may also be perfectly acceptable. In the case of double-stranded oligonucleotide drug substances and drug products, provided intact masses of the individual single strands are measured routinely before annealing, drug substance and drug product identity testing can be reduced to an intact mass measurement coupled with a confirmation of the double-stranded nature of the sample (non-MS-based strategies for double-stranded drug products may also be acceptable).
We believe that the risk assessment presented above should apply broadly, and, therefore, that the analytical approaches outlined will be sufficient to confirm the identity of most oligonucleotides. Nevertheless, before implementation, we strongly recommend that sponsors of therapeutic oligonucleotides conduct and document a similar risk assessment to check for potential gaps not covered by the general example presented here. At the very least, circumstance-specific risk assessments will likely help sponsors select among the various secondary drug substance identity test methods that accompany intact mass measurement (which is a powerful test for all oligonucleotide drug substances). For example, in oligonucleotides that contain an equal number of phosphorothioate and phosphate diester linkages, the failure mode of swapping the positions of oxidizer and sulfur transfer tanks (Fig. 1) results in an isobaric oligonucleotide of the same base sequence as the intended oligonucleotide. The sponsor might then use this circumstance-specific knowledge to decide among a number of secondary identity testing methods by determining experimentally which most reliably distinguishes the intended oligonucleotide from the backbone isomer.
The consequences of dosing a patient with an unintended oligonucleotide are severe. However, we are confident that the integrated risk assessment discussed here will allow sponsors and regulators to communicate a common understanding of the issues. In turn, this should result in a consistent, science-based approach to identity testing of therapeutic oligonucleotides that will facilitate access to safe, efficacious, and potentially life-changing medicines.
Footnotes
Acknowledgments
The authors acknowledge Tracey Reigle (Ionis Pharmaceuticals, Inc.) for graphics art assistance, and Alfred Ross (F. Hoffmann—La Roche Ltd.) and David Dias (AstraZeneca) for NMR measurements and spectrum interpretation.
Author Disclosure Statement
The authors declare no further interest beyond their employment by the companies as indicated.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.