
Abstract
Hyperammonemia is a dangerous life-threatening metabolic complication characterized by markedly elevated ammonia levels that can lead to irreversible brain damage if not carefully monitored. Current pharmacological treatment strategies available for hyperammonemia patients are suboptimal and associated with major side effects. In this study, we focus on developing and evaluating the in vivo delivery of novel DNA-encoded glutamine synthetase (GS) enzymes for the treatment of hyperammonemia. Direct in vivo delivered DNA-encoded GS enzyme was evaluated in ammonium acetate-induced hyperammonemia and thioacetamide-induced acute liver injury (ALI) models in C57BL/6 mice. In ammonium acetate-induced hyperammonemia model, we achieved a 30.5% decrease in blood ammonia levels 15 min postadministration of ammonium acetate, with DNA-encoded GS-treated group. Significant increase in survival was observed in ALI model with the treated mice. A comparison of the secreted versus intracellular DNA-encoded GS enzyme demonstrated similar increases in survival in the ALI model, with 40% mortality in the secreted enzymes and 30% mortality in the intracellular enzymes, as compared with 90% mortality in the control group. Direct in vivo delivery of DNA-encoded GS demonstrated important ammonia-lowering potential. These results provide the initial steps toward development of delivered DNA as a potential new approach to ammonia-lowering therapeutics.
Introduction
Hyperammonemia is a dangerous complication that can manifest as a result of inborn urea cycle enzyme deficiency disorders, inborn errors of branched amino acid metabolism, acute or chronic liver diseases, transient hyperammonemia of the newborn, renal disease, trauma, and gastrointestinal bleeding, among other causes [1–3]. Ammonia is a potent neurotoxin, which if highly elevated can lead to irreversible brain damage. In normal physiological conditions, the urea cycle in the liver converts NH4+ to urea which undergoes renal excretion. However, in cases of liver damage or failure, the elevated circulatory ammonia levels can result in hepatic encephalopathy, manifested by elevated intracranial hypertension and brain edema, culminating in coma and even death [4,5]. Healthy human adults have blood ammonia levels below 50 μM [6]. Different pathological conditions result in different levels of plasma ammonia, some requiring immediate hemodialysis. Urea cycle disorders can result in plasma ammonia levels ranging from 100 μM to >1,000 μM. Liver or renal failure conditions generally present with elevated ammonia levels between 50 and 500 μM [7–9].
Today's pharmacotherapeutic strategies available for treating hyperammonemia in pediatric or adult patients have been inefficient at bringing about rapid and robust decrease in ammonia levels. Current treatment strategies for pediatric hyperammonemia include protein restriction (restrict nitrogen intake), administration of sodium benzoate (to remove nitrogen by forming conjugates with glycine and glutamine), arginine and ornithine (to stimulate urea cycle), disaccharides and antibiotics (to reduce gastrointestinal production of ammonia), exchange transfusion, peritoneal dialysis, or hemodialysis [1,10,11]. Current treatment guidelines require that patients with ammonia levels above 500 μM should undergo hemodialysis. Patients with ammonia levels below 500 μM can be treated with pharmacotherapeutics, which to date have been inefficient to bring about sufficient decrease. Preparations for hemodialysis are made when ammonia levels reach close to 500 μM threshold and cannot be adequately controlled. Hemodialysis has been by far the most efficient treatment for hyperammonemia in both pediatric and adult patients; however, it is riskiest in terms of the complications that can arise [1,12]. Development of novel potent ammonia-lowering therapeutics is essential for the treatment of hyperammonemia, and in particular, to reduce ammonia levels and prevent elevation above the 500 μM threshold, to avoid requirement for hemodialysis, which bears risk of potentially fatal complications and is not available in all communities.
Typically, ammonia is detoxified by the glutamine synthetase (GS) enzyme present in liver, muscle, kidneys, and brain, as well as by the hepatic urea cycle [13]. GS is a key enzyme in ammonia detoxification, which becomes even more important in the cases of inborn errors of metabolism or liver failure [13]. The first demonstration of gene therapy to treat hyperammonemia was performed in 2015 by Torres-Vega et al. [14], who introduced GS gene using baculovirus delivery system into the skeletal muscle of rats for treatment of acute hyperammonemia, achieving a significant 351 μM reduction in serum ammonia levels in rats. GS gene therapy could be very valuable for treatment of hyperammonemia. One form of direct gene delivery for therapeutic GS is to examine the direct local electroporation-enhanced, synthetic DNA-encoded therapeutic protein expression platform. In this study, the in vivo CELLECTRA electroporation device is used to induce low-voltage electropermeabilization to efficiently deliver the plasmid DNA to the skeletal muscle [15,16]. This is a more cost-effective approach than the recombinant protein-based systems, which have both cost and manufacturing challenges [16,17]. The cost-effectiveness of this technology could also alleviate disparity across economic and racially disadvantaged groups in the United States. There are a number of advantages to using this technology in addition to cost effectiveness, and these include the ease of manipulation, stability of product, repeat administration, lack of antivector response after repeat treatments, reduced number of administrations, and the lack of a requirement for cold-chain distribution due to the inherent stability of DNA biologicals [18–20].
Our objective in this study, was to develop an ammonia-lowering therapeutic that could be administered to hyperammonemic patients, who have plasma ammonia levels below 500 μM threshold, to prevent the escalation in plasma ammonia levels. In this study, we developed a novel DNA-encoded GS enzyme therapy using direct in vivo gene delivery for the treatment of hyperammonemia. Murine GS enzyme sequence was codon optimized to enhance GS mRNA stability and translation efficiency in mice. Two enzyme constructs were developed, one with the native leader sequence of the enzyme for intracellular expression, and the other with highly efficient secreted IgE leader sequence for secretion of GS enzyme. The developed genes were cloned into a pVax-1 mammalian vector and evaluated for expression both in vitro and in vivo. Initial characterization showed that robust enzyme activity was detected in the in vitro studies, which showed a significant reduction in ammonia in an in vitro hyperammonemia model. We next examined the effects of delivery of the plasmids on the therapeutic GS enzyme activity in vivo through an ammonium acetate-induced hyperammonemia model in C57BL/6 mice. For these studies we delivered the constructs 3 days before ammonium acetate administration. We observed that the constructs demonstrated a significant drop in ammonia levels as soon as 15 min after ammonium acetate administration. Furthermore, efficacy of the therapeutic enzymes was evaluated in an acute disease setting, a thioacetamide-induced acute liver injury (ALI) model. Substantial reduction in mortality was observed with GS-treated mice. These results demonstrate the potential of these simple synthetic DNA-encoded GS enzymes for treatment of hyperammonemia.
Materials and Methods
Cell culture
Human embryonic kidney (HEK) 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco-Life Technologies) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies).
GS plasmid design and construction
Two GS constructs were developed: (1) intracellular murine GS, and (2) secreted murine GS. The transgenes were synthesized and subcloned into the corresponding restriction enzyme site in pVax-1 mammalian expression vector. For the secreted GS enzyme, an highly efficient IgE leader sequence was inserted on the 5′ end of GS sequence to improve protein translation. Human cytomegalovirus immediate-early promoter in the pVax-1 vector was used to drive the expression of the enzymes.
In vitro transfection of GS plasmids
HEK293T cells were transfected with GS plasmids using Lipofectamine LTX reagent (Thermo Fisher Scientific) and following the manufacturer's protocol. Briefly, in a 12-well plate, 5 × 105 cells were seeded and incubated in a 37°C incubator with 5% CO2. Cells were transfected with 1 μg GS plasmid at 70% confluence and incubated for a maximum of 72 h. Enzyme expression and activity following transfection was analyzed by western blot, enzyme-linked immunosorbent assay (ELISA), enzyme activity assay, and immunofluorescence microscopy.
Western blot and ELISA analysis
A denaturing and native western blot analysis was performed to evaluate the size and the oligomeric nature of GS. Western blot analysis will be performed on the cellular supernatant and lysates from 293T transfected cells at 24-, 48-, and 72-h time points.
Denaturing western blot was performed by running the supernatant of the transfected cells in precast NuPAGE 10% Bis-Tris gels (Invitrogen) under reducing conditions. Gels were transferred to polyvinylidene difluoride transfer membranes (Millipore). The membranes were blocked in 3% nonfat dry milk in phosphate-buffered saline (PBS) with 0.1% Tween 20 (Sigma-Aldrich). Native western blot was performed by running the supernatant of the transfected cells in precast NativePAGE 3%–12% Bis-Tris gels (Invitrogen) following the manufacturer's protocol. The membrane was stained with anti-mouse GS primary antibody (Abcam) and an anti-rabbit IgG-horseradish peroxidase (HRP) secondary antibody (Abcam).
GS enzyme was quantified by ELISA. ELISA MaxiSorp plates (Thermo Fisher Scientific) were coated with samples and the recombinant GS protein (LifeSpan BioSciences) standards, and incubated overnight at 4°C. Plates were blocked in 10% FBS in PBS for 1 h at room temperature (RT). After washing the plates, anti-mouse GS primary antibody (Abcam) was added for 1 h at RT, followed by anti-rabbit IgG-HRP secondary antibody (Abcam) for 1 h at RT. SigmaFast OPD (Sigma-Aldrich) substrate solution was used for sample detection. Optical density was measured at 450 nm using a GloMax 96 Microplate Luminometer (Promega).
Enzyme activity assay and ammonia quantification
GS enzyme activity was measured using the GS Microplate Assay Kit (Cohesion Biosciences) on supernatant and lysate samples of transfected 293T cells, according to the manufacturer's protocol. At different time points postadministration of exogenous ammonia, the ammonia levels were quantified using the Colorimetric Ammonia Assay Kit (Abcam) on cellular supernatant and lysate samples, according to the manufacturer's protocol.
Immunofluorescence analysis
The 293T cells were transfected with GS plasmids using Lipofectamine LTX reagent as described above. Forty-eight hours posttransfection, cells were washed in PBS and fixed in 4% paraformaldehyde for 10 min at RT. Cells were then washed and incubated with anti-mouse GS primary antibody, and anti-rabbit IgG-fluorescein isothiocyanate (FITC) (Abcam) for 1 h. After washing, cells were stained with DAPI nuclear stain (Thermo Fisher Scientific) and imaged on Nikon TE2000 Inverted Microscope Imaging System.
Immunohistochemistry of mouse muscle
Immunohistochemistry was performed by The Wistar Institute Histotechnology Facility. Briefly, mouse anterior tibialis muscle sections were resected and embedded in paraffin. Paraffin-embedded samples were treated with antigen retrieval reagent and deparaffinized. Slides were then fixed with acetone and washed with PBS, followed by blocking and staining the sections with anti-mouse GS primary antibody (Abcam) and anti-rabbit IgG-HRP secondary antibody (Abcam). Hematoxylin and Eosin (H&E) staining, and Masson's Trichrome staining was performed on the liver sections for visualization of severity of liver damage induced by the ALI.
In vivo DNA-encoded GS plasmid administration
Evaluation of DNA-encoded GS was carried out in 6- to 8-week-old C57BL/6J wild-type mice. Animal experiments were carried out in accordance with the guidelines of the National Institutes of Health (NIH) and The Wistar Institute Institutional Animal Care and Use Committee (IACUC). Mice were injected with GS plasmid DNA in the tibialis anterior or quadriceps muscles. In vivo electroporation was carried out using a CELLECTRA adaptive constant current EP device (Inovio Pharmaceuticals). Triangular 3-electrode arrays consisting of 26-gauge solid stainless steel electrodes were used to deliver square-wave pulses. Two constant current pulses were delivered at 0.1 Amps for 52 microseconds/pulse, separated with a 1 s delay between pulses. Mice were given a dose of 100–300 μg plasmid DNA resuspended in water. To enhance their intramuscular distribution, the plasmids were coformulated with recombinant hyaluronidase. A 30 μL volume was used at each injection site. As a negative control for DNA-encoded GS, pVax-1 empty vector was used. Blood was collected by submandibular bleeding for GS quantification and enzyme activity analysis.
Ammonium acetate-induced hyperammonemia model
Female C57BL/6 mice (n = 5 mice per group) were administered 200 mg/kg ammonium acetate (Sigma-Aldrich) through intraperitoneal injection at day 3 posttreatment with 200 or 300 μg of DNA-encoding GS plasmids or control pVax-1 plasmids. Ammonia levels were measured at 0 and 15 min after ammonium acetate administration. Study was performed in duplicates.
Thioacetamide-induced ALI model
For induction of thioacetamide-induced ALI in mice, female C57BL/6 mice (n = 10 mice per group) were injected by intraperitoneal injection either a high dose of 250 mg/kg or a low dose of 150 mg/kg in 0.9% NaCl on days 0 and 1. The mice were administered with 200 or 300 μg of DNA-encoding GS plasmids or control pVax-1 plasmids, 2 days before thioacetamide administration. Mouse survival was monitored either over a short-term 5-day period or long-term 3-month period. The mice were monitored regularly using neurobehavior and pain perception tests. Study was performed in duplicates.
Clinical behavior and pain perception scoring system
The clinical behavior scoring system was used to measure the severity of the ALI, with score of 0 indicative of normal behavior and score of 4 as lack of reaction to pain stimuli and no righting reflex. In accordance with IACUC regulations, mice were euthanized at a clinical behavior score of 4. The clinical scoring system was performed according to Farjam et al. [21]. Score: 0-normal behavior; 1-mild lethargy; 2-decreased motor activity, poor gesture control, diminished pain perception; 3-Severe ataxia, no spontaneous righting reflex; 4-No righting reflex, no reaction to pain stimuli. Pain perception was scored by the toe pinch and the tail pinch tests. The following scoring system was used to distinguish the level of pain perception. Three pinches were performed per test. Toe pinch score: 0-none; 1-slight withdrawal; 2-moderate rapid withdrawal, not brisk; 3-brisk, rapid withdrawal; 4-very brisk, with repetitive extension and flexion; tail pinch score: 0-no response; 1-very slight movement; 2-slight bite, escape; 3-moderate bite, escape; 4-vigorous biting and escape; 5-extremely vigorous biting and escape.
NanoString nCounter mRNA profiling
mRNA expression profile analysis was performed using NanoString nCounter mouse neuroinflammation panel (NanoString Technologies). Total RNA was extracted from brain cortex tissues from GS and control pVax-1-treated C57BL/6 mice (n = 3 mice per group). RNA extraction was performed by the Wistar Genomics Core. RNA samples were profiled according to standard NanoString protocols on a NanoString nCounter Analysis System operated by the Wistar Genomics Core. Data were analyzed using the NanoString n-Solver software.
Statistics
Experimental data were analyzed using one-way analysis of variance and log-rank tests. Differences were deemed significant at P-values <0.05. All graphs were prepared using GraphPad Prism 6 (GraphPad Software).
Results
Expression and functional activity of DNA-encoded GS in vitro
Plasmid DNA were designed to encode both secretable and intracellular forms of murine GS. For one construct a highly efficient IgE leader sequence was inserted on the 5′ end of a secretable form of GS sequence (Fig. 1A). Enzyme expression was evaluated by immunofluorescence microscopy (Fig. 1B) on 293T cells transfected with the secretable GS or pVax-1 control plasmids, and stained with anti-mouse GS antibody. Production of enzyme was confirmed in vitro through transient transfection of 293T cells. Denaturing (Fig. 1C) and Native (Fig. 1D) gel western blot analysis was performed on the cellular supernatant from 293T cells transfected with secretable GS. A band near the 38 kDa marker corresponding to the monomer of GS enzyme was observed with the denaturing western blot under reducing conditions. Native PAGE western blot analysis showed bands ranging from 420 to 720 kDa markers. Mouse GS is thought to be homooctameric [22] as compared with the human counterpart, which is homodecameric [23]. To evaluate the capability of the designed therapeutic enzymes in metabolizing ammonia, an in vitro hyperammonemia model (Fig. 1E, F) was performed. The 293T cells were transfected with a secretable GS plasmid DNA. Twenty-four hours later, cells were incubated with 100 μM ammonium chloride. Ammonia levels were quantified at different time points using a colorimetric ammonia assay. Compared with media alone with the exogenous ammonium chloride, a significant 37.4% drop in ammonia levels was observed at 48 h in the supernatant from the GS-transfected cells.
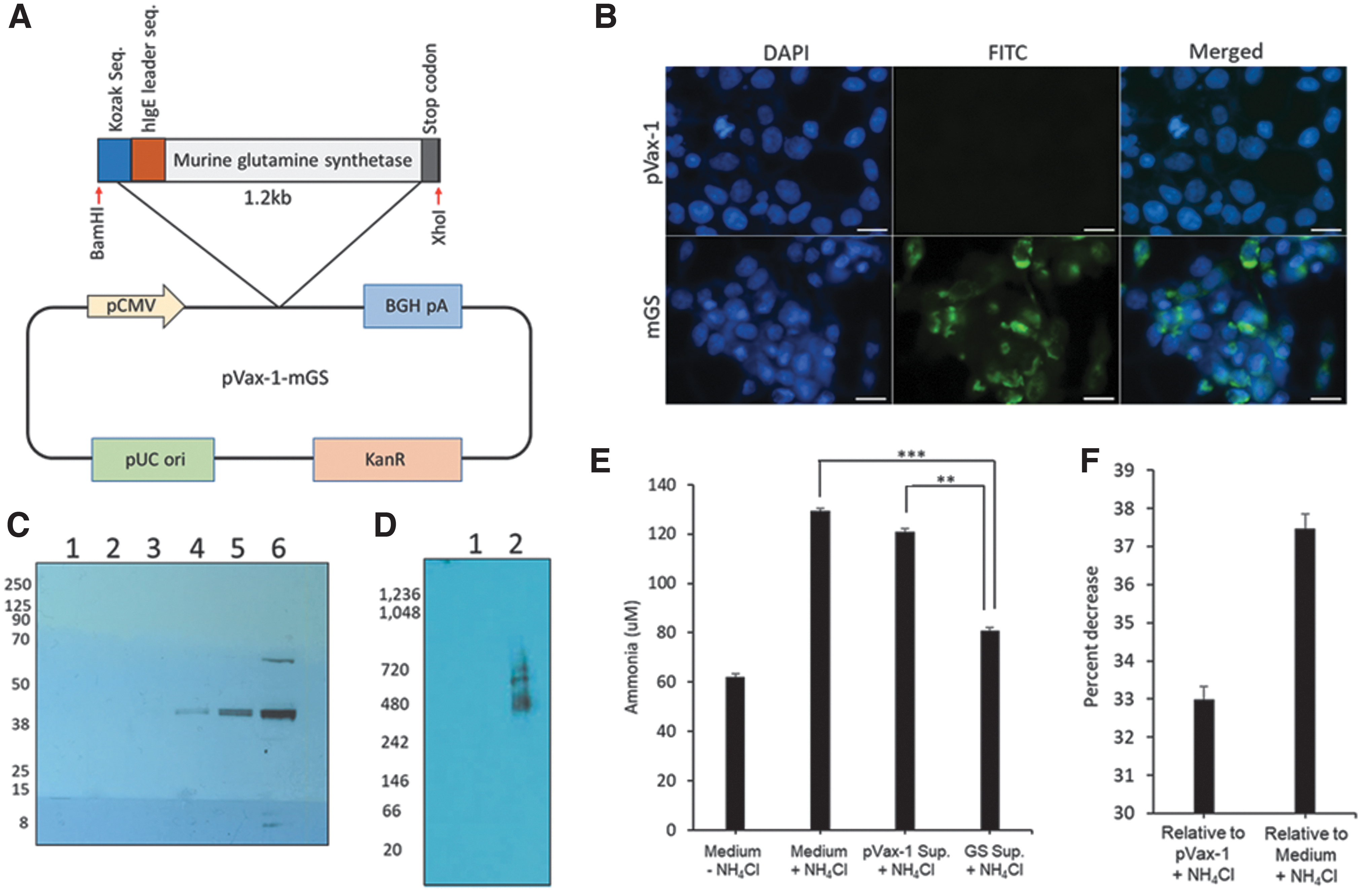
In vitro characterization of DNA-encoded murine GS.
Expression kinetics and sustainability of DNA-encoded GS enzymes in vivo
Intramuscular expression of secreted GS was evaluated in 6–8-week-old female C57BL/6 mice. An in vivo electroporation device (Fig. 2A) was used for delivery of GS plasmids into skeletal muscle for in vivo enzyme expression. GS enzyme activity analysis of the treated muscle tissue demonstrated increasing enzyme activity from 0.07 U/mg at day 0 to 0.19 U/mg at day 5 (Fig. 2B). Quantitative ELISA for quantification of intramuscular GS enzyme showed around 0.83 ng/mg at day 0 compared with 9.63 ng/mg at day 5 (Fig. 2C). Enzyme expression was also confirmed by immunohistochemistry of the mouse muscle tissue. As shown in Fig. 2D, immunostaining with anti-mouse GS antibody-HRP indicated positive browning in the mouse tibialis anterior muscle section, compared with slight browning in the control pVax-1 muscle sections. The natural skeletal muscles express GS enzyme, which can be observed through the enzyme activity assay, ELISA, and IHC, however, in these studies, we confirmed that the expression was much stronger in the CELLECTRA-delivered muscle sections.
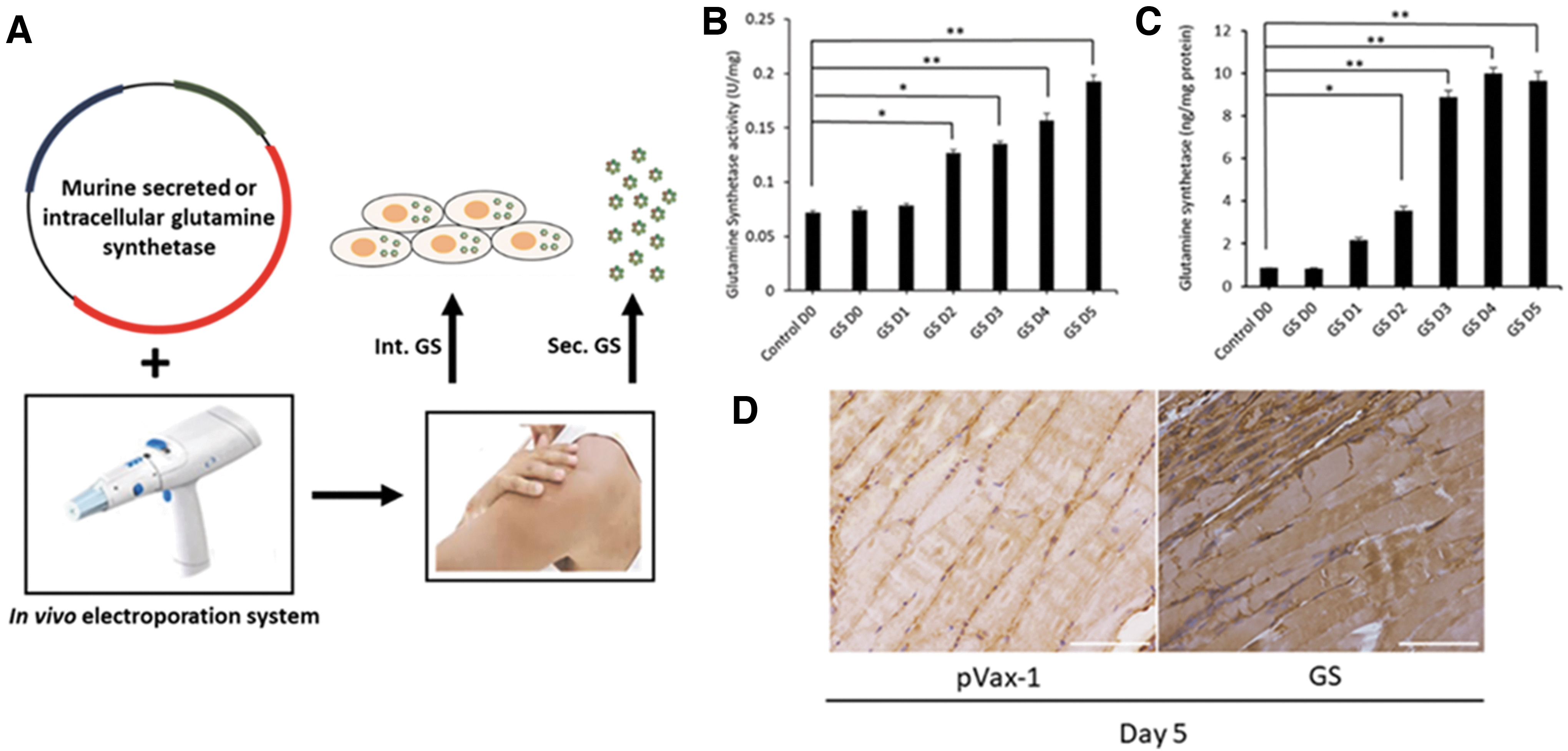
Intramuscular expression analysis of DNA-encoded GS.
The expression level and duration of the DNA-encoded secreted GS enzyme were evaluated in the serum of 6–8-week-old female C57BL/6J mice. Two hundred micrograms of plasmid DNA was injected intramuscularly into the tibialis anterior muscle, followed by electroporation (IM-EP). Enzyme activity and quantitative ELISA were performed on mouse serum samples taken periodically for 42 days. Upon single intramuscular administration (Fig. 3A, B) of the GS plasmids, greatest enzyme activity and enzyme levels in the serum were observed on days 3 and 5. Sequential administration (Fig. 3C, D) of the plasmids on days 0, 5, and 10, enabled longer duration of the enzymes up to day 14. The duration of the enzyme expression in the serum is significantly higher than intravenously administered recombinant murine GS enzyme (1 mg/kg dose), which drops drastically in the serum after 5 min following administration (Fig. 3E). This highlights the sustainability of GS expression over long periods. Further improvements in durability of GS expression was observed when doses were sequentially administered over several days.
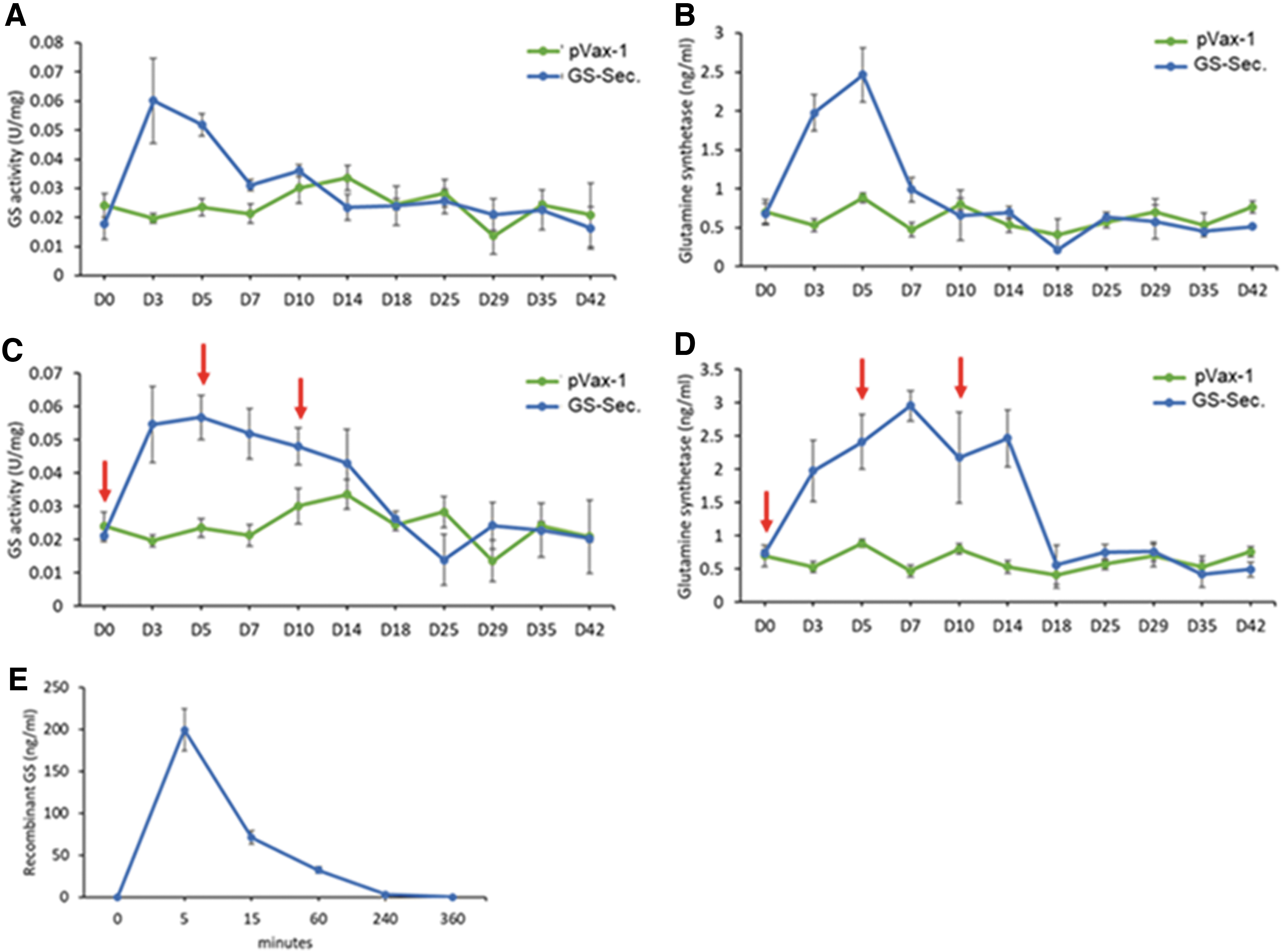
In vivo expression and kinetics of DNA-encoded secreted GS.
Reduction in ammonia by DNA-encoded GS in an ammonium acetate-induced hyperammonemia
To evaluate ammonia-lowering capability of DNA-encoded GS, an acute hyperammonemia model was next employed. In this model, intravenous administration of ammonium acetate brings about rapid and transient increase in plasma ammonia levels. This model provides a small time-window to evaluate the possible ammonia-lowering therapeutic effect. Ammonium acetate-induced hyperammonemia model used, studied the efficacy of the DNA-encoded therapeutic GS enzymes in 6–8-week-old female C57BL/6 mice. The mice were administered 200 mg/kg ammonium acetate at day 2 posttreatment with 200 or 300 μg DNA-encoding secreted GS plasmids. Ammonia levels were measured 15 min after ammonium acetate administration. We observed a 30.5% decrease in ammonia levels after 15 min postadministration of ammonium acetate, in the 300 μg plasmid dose-secreted GS-treated group (Fig. 4). All animals clear the levels of ammonium acetate after longer time periods. These results demonstrate for the first time a potential of DNA-encoded GS enzyme therapy for targeting hyperammonemia in vivo.

Evaluation of DNA-encoded secreted GS in ammonium acetate-induced hyperammonemia. C57BL/6 mice were administered with 200 mg/kg ammonium acetate through intraperitoneal injection on day 3 posttreatment with DNA-encoded GS.
Reduced mortality by DNA-encoded GS in ALI model
To further evaluate the efficacy of the therapeutic enzymes in an acute disease setting, a thioacetamide-induced ALI model was used. The major cause of mortality from ALI is hepatic encephalopathy. In this model, mice can present with drastically elevated ammonia levels, neurobehavioral deficits, and increased mortality. Both high dose (250 mg/kg) and low dose (150 mg/kg) of thioacetamide administration were used to evaluate the effect of therapeutics in different severities of ALI. Thioacetamide-induced ALI was performed in 6–8-week-old female C57BL/6 mice by treating mice with two intraperitoneal injections of thioacetamide (either 150 or 250 mg/kg). In the high-dose thioacetamide-induced ALI (Fig. 5), the pVax-1 control group had a 60% mortality at day 2, and a 100% mortality at day 4 post-thioacetamide administration, however, in the GS-treated mice with 300 μg plasmid DNA dose, there was zero mortality on day 2, and a 50% mortality at the end of study on day 5. Two hundred micrograms of plasmid DNA dose was not as efficient as the higher dose, however, it still reduced mortality compared with the pVax-1 control group. A low-dose 150 mg/kg thioacetamide-induced ALI model was used to evaluate the therapeutic efficacy of the secreted as well as the intracellular forms of GS enzyme. A 40% mortality was observed with the therapeutic GS enzymes, as compared with 90% mortality with the pVax-1 control group, on day 5 (Fig. 6). No difference in mortality was observed between the secreted or the intracellular forms of GS enzymes. A clinical behavior scoring system along with toe-pinch and tail-pinch pain perception tests were used to monitor the neurobehavior status of mice. Neurobehavioral and pain perception deficits were significantly lower in the GS-treated mice. Long-term study of the low thioacetamide dose ALI model was performed to evaluate efficacy of the enzyme therapeutics over a 2-month period (Fig. 7A). Deaths of animals were only observed within the initial 5 days post-thioacetamide administration. No increases in mortality was observed over an extended period, possibly due to liver regeneration. At the end of the 2-month period, pVax-1 control group had a 90% mortality as compared with the secreted GS groups with 40% mortality and intracellular GS with 30% mortality.

Evaluation of DNA-encoded secreted GS in high-dose thioacetamide-induced ALI mouse model. C57BL/6 mice were administered 250 mg/kg thioacetamide through intraperitoneal injection on days 0 and 1, with DNA-encoded secretable GS administered on day 2.

Evaluation of DNA-encoded secreted and intracellular GS in low-dose thioacetamide-induced ALI mouse model. C57BL/6 mice were administered 150 mg/kg thioacetamide through intraperitoneal injection on days 0 and 1, with 300 μg dose of DNA-encoded secretable GS administered on day 2.
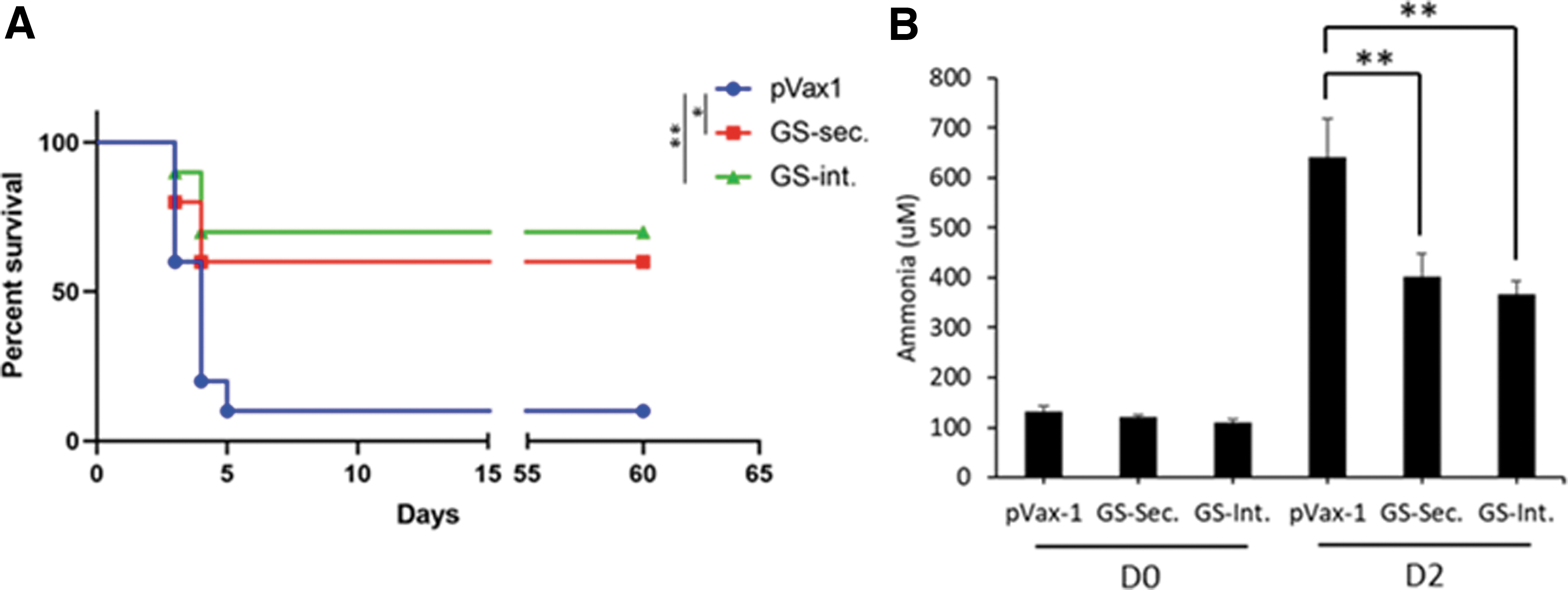
Long-term evaluation of DNA-encoded secreted and intracellular GS in low-dose thioacetamide-induced ALI mouse model. C57BL/6 mice were administered 150 mg/kg thioacetamide through intraperitoneal injection on days 0 and 1, with 300 μg dose of DNA-encoded secretable GS administered on day 2. The mice were evaluated for a total of 60 days.
On day 2, following the thioacetamide-induced ALI, the plasma ammonia levels were markedly elevated, which were significantly reduced with the GS treatments. Plasma ammonia levels were reduced from a mean of 641.1 μM ammonia in the pVax-1 control group to 401.2 μM ammonia for GS-Sec. group and 366.3 μM ammonia for GS-Int.-treated mice (Fig. 7B). The severity of the ALI was monitored by serum aspartate aminotransferase/alanine aminotransferase (AST/ALT) aminotransferase enzyme levels (Supplementary Fig. S1A, B). AST/ALT levels were markedly elevated on day 2 as a result of the ALI, which corresponded with H&E staining showing necrotic cores, as well as Trichrome staining showing increased collagen deposition and scarring of the liver tissue, demonstrating successful induction of ALI in all groups of animals (Supplementary Fig. S1C).
Molecular pathways underlying changes in neuroinflammation following treatment with DNA-encoded GS
To investigate changes in molecular pathways associated with neuroinflammation in the brain tissue with or without treatment with GS enzyme, an mRNA expression profile analysis was performed using NanoString nCounter mouse neuroinflammation panel (Fig. 8). In this study, we can identify minute changes in neuroinflammation pathway between treated and untreated mice. Hierarchical clustering (Fig. 8A) showed significant changes in mRNA profiles between pVax-1 control mice compared with the GS-treated mice in the low-dose thioacetamide-induced ALI mode. The clustering showed that both the secreted and intracellular GS treated mice grouped together showing similar changes in gene expression, as compared with the pVax-1-treated mice that clustered apart. As shown in the volcano plots (Fig. 8B, C) comparing GS-treated groups versus pVax-1 control group, the genes that were significantly downregulated in the GS-treated groups were Ttr, Agt, Slc17a6, Tspan18, Fbln5, Amigo2, Hsd11b1, and Cd36. Agt (angiotensin gene) is expressed by astrocytes and some neurons in the brain, and plays a role in regulating blood pressure and electrolyte balance [24,25]. Ttr (transthyretin) expression has been reported to increase in response to CNS injury, and could play a role in neuroprotection [26,27]. SlC17a6 gene that encodes for vesicular glutamate transporter 2 is involved in various functions in neurocircuitry [28]. Most of these genes play neuroprotective roles and are upregulated during injury. It is possible that the GS enzyme through reducing the neuroinflammation or CNS damage in the brain is reducing their upregulation compared with the hyperammonemic pVax-1 group. Genes that were significantly upregulated in the GS-treated mice include Blnk and Top2a. Blnk (B cell linker) plays a role in the adaptive immune response [29]. Top2a (DNA topoisomerase 2 alpha) plays a role in controlling the topological state of genomic DNA [30]. Notably, both the secretable and intracellular GS enzymes preformed similarly as shown by the heat map and volcano plots. These results support the significant differences in genes associated with neuroinflammation that were altered in vivo as a result of treatment by the therapeutic DNA-encoded GS enzymes.
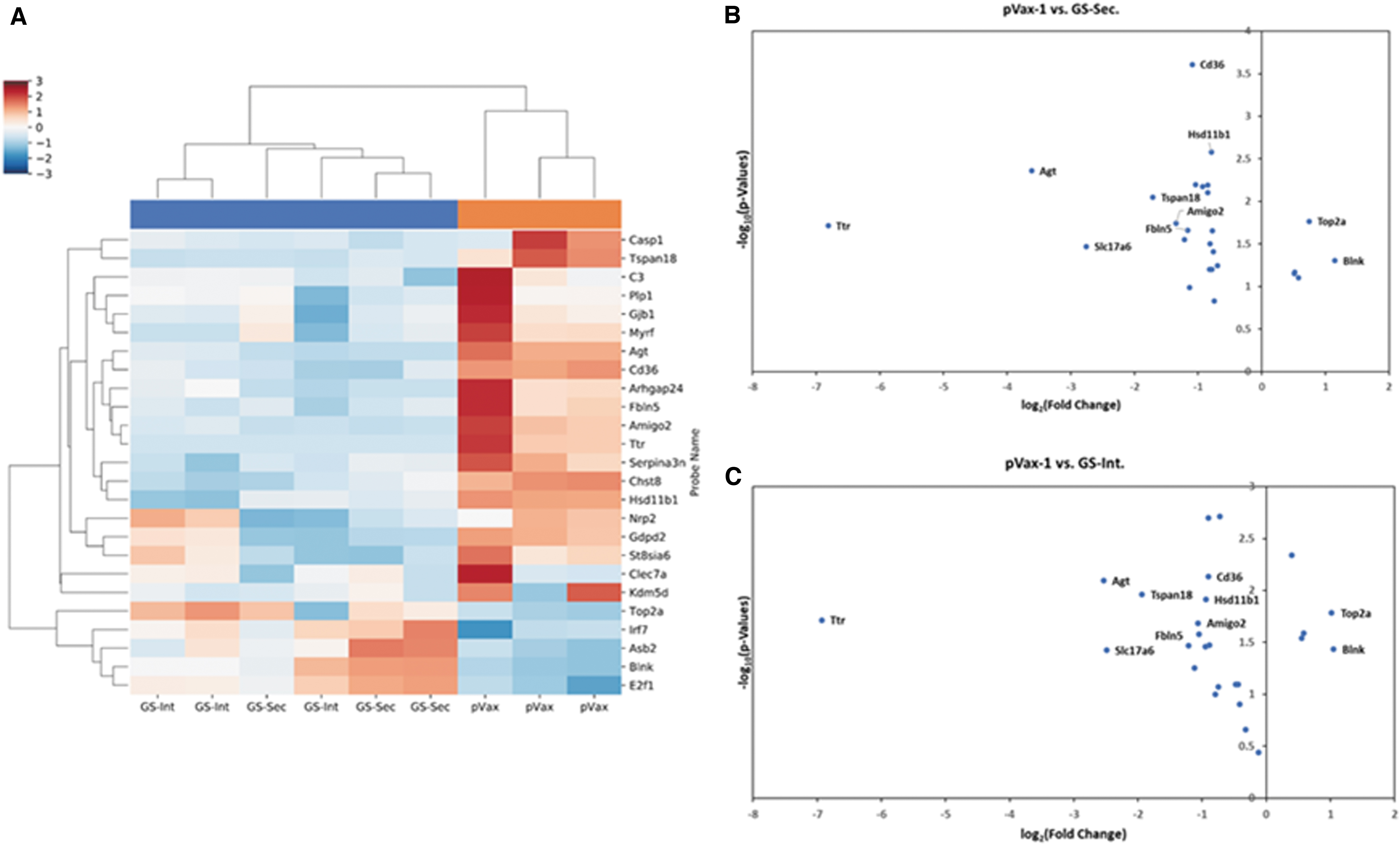
Changes in mRNA expression of neuroinflammation-associated gene in the brain cortex of mice with low-dose thioacetamide-induced ALI. C57BL/6 mice were treated with DNA-encoded GS or pVax-1 control plasmids on day 2. Brain cortex tissues were harvested on day 2 post-thioacetamide administration. Nanostring gene expression analysis was performed using mouse neuroinflammation panel.
Discussion
Hyperammonemia is a clinical condition of ammonia intoxication that can result from a range of hereditary or acquired pathological conditions. Medical emergency presents due to neurological impairment resulting from ammonia toxicity, which could lead to potentially fatal outcome. Today's pharmacotherapeutic strategies available for hyperammonemia patients are inefficient at bringing about rapid and potent reduction in ammonia levels, and most associated with adverse side effects. Generally, pharmacotherapy is administered at ammonia levels below 500 μM, and anything above requires rapid hemodialysis to avoid risk of irreversible brain damage. Hemodialysis has been by far the most efficient treatment for hyperammonemia in both pediatric and adult patients; however, it is riskiest in terms of the complications that can arise [1,12]. Regular hemodialysis machines are not designed for pediatric use. Specialized centers are required for performing neonatal or pediatric hemodialysis. Young age has been a main factor in poor outcome in children with hemodialysis [31]. Development of novel potent ammonia-lowering therapeutics is essential for treatment of hyperammonemia, and in particular to reduce or eliminate the reliance on hemodialysis, which bears risk of potentially fatal complications and is not available in all communities.
In 2015, Torres-Vega et al. [14] reported on the use of baculoviral vector to deliver GS intramuscularly to the hind legs of rats to treat acute hyperammonemia. Ammonium acetate-induced hyperammonemia model was used to evaluate the efficacy at reducing plasma ammonia levels. Fifteen minutes after administration of 200 mg/kg ammonium acetate, the plasma ammonia levels were reduced by a significant 66% in the treated group, corresponding to a 351 μM ammonia reduction. The same group, recently evaluated their GS baculovirus platform in hyperammonemic bile duct-ligated rat model to evaluate the therapeutic in chronic liver failure [32]. The resulting hyperammonemia was controlled by the baculovirus-delivered GS administration in the muscle, with a significant 56.2% reduction in the plasma ammonia levels on day 3 posttreatment administration. This is a major accomplishment for treatment of both acute and chronic hyperammonemic pathological conditions. It should be noted that baculovirus is an immunogenic vector, and therefore vulnerable to immune-induced suppression of transgene expression. Other nonimmunogenic delivery systems such as DNA electroporation could provide longer lasting expression of the GS enzyme. In our studies, secreted GS was detectable in the plasma until day 5 with single-dose administration, and day 14 with sequential dose administration. Further dosing and sequential administration studies are required to evaluate the long-term expression of both the secreted and intracellular GS enzymes. Soria et al. [33] utilized helper-dependent adenovirus-based delivery of murine GS for hepatic enhancement of ammonia detoxification. In an acute hyperammonemia model in wild-type mice, using an ammonium chloride challenge, a 39% decrease in serum ammonia levels was seen after 30 min in the treatment group. The treatment was also able to protect from ammonia challenge in a transgenic carbamoyl phosphate synthetase 1 (Cps1)-deficient mice, which shows their potential for treatment of hyperammonemia in urea cycle disorders. Another interesting area of research that has been used to treat hyperammonemia is through the use of engineered gut microbiota. Shen et al. [34] engineered murine gut microbiota to have reduced urease gene content to decrease ammonia production. In a high-dose thioacetamide-induced ALI, all the control rats died after around 50 h, as compared with the treatment group, which had around 30% survival at 170 h. In our ALI model using high-dose (250 mg/kg) thioacetamide, we achieved a 50% survival by day 3 using the DNA-encoded GS. Both gene therapy and microbial engineering offer promising therapeutic alternatives for treatment of acute and chronic hyperammonemia.
DNA-encoded electroporation-enhanced delivery is an expression platform that offer nonimmunogenic delivery of plasmid DNA for in vivo protein expression. In this study, in vivo CELLECTRA electroporation device is used to induce low-voltage electropermeabilization to efficiently deliver the plasmid DNA to the skeletal muscle. This provides a more cost-effective approach than the recombinant protein-based therapy, which has both cost and manufacturing challenges. The advantages of DNA-encoded expression platform include cost effectiveness, ease of manufacturing, stability, and no requirement for a cold chain and rapid development and engineering. This platform has been used by our group for DNA-encoded monoclonal antibody (DMAb) expression in vivo and has shown protective efficacy against several viral pathogens, including HIV, Flu, MERS, and Zika among others, as well as therapeutic efficacy for the treatment of hypercholesterolemia using DNA-encoded anti-PCSK9 antibodies [18–20,34–42]. Furthermore, with respect to nucleic acid immunogenicity, no anti-DNA immune responses have been reported, and DNA platforms are reported to be very tolerable and safe [43,44].
In this study, we evaluated the efficacy of the DNA-encoded GS enzyme platform for treatment of hyperammonemia. In an in vitro hyperammonemia model using exogenously added ammonium chloride to 293T-transfected cells, a significant 37.4% drop in ammonia levels was observed at 48 h. The efficacy of the DNA-encoded enzyme was evaluated in vivo in C57BL/6 mice using an ammonium acetate-induced transient hyperammonemia model. The 300 μg plasmid DNA dose of the enzyme was able to achieve a 31.2% reduction in plasma ammonia levels at 15 min postammonium acetate administration. To evaluate the efficacy of the enzyme further in a clinically relevant hepatic encephalopathy model, a thioacetamide-induced ALI was used in C57BL/6 mice. In a high-dose thioacetamide (250 mg/kg)-induced ALI model, a 300 μg plasmid dose of GS was able to result in a 50% survival at day 5, as compared with the control group, which had a 100% mortality on day 4. Survival was also evaluated in a low-dose thioacetamide (150 mg/kg)-induced ALI. In this study, comparing the secreted versus intracellular GS enzymes at 300 μg plasmid DNA dose demonstrated equal protection against hyperammonemia with 60% survival, as compared with the control group with 10% survival at day 5. Correspondingly, the neurobehavior and pain perception of the hyperammonemic mice significantly improved with the therapeutic enzymes. The study was repeated over a 2-month period with the intracellular GS performing slightly better, with survival of 70% for GS-Int., 60% for GS-Sec., and 10% for pVax-1 control mice at day 60. As observed in these studies, a single intramuscular administration of GS plasmid DNA was able to lead to significant reduction in ammonia levels in ammonium acetate-induced hyperammonemia model, as well as a substantial decrease in mortality in an ALI model. NanoString analysis of neuroinflammation in the brain cortical sections in the ALI model, showed that with the expression level of neuroprotective gene (such as Amigo2, Slc17a6, Ang, Ttr, and others) were higher in the pVax-1 control group, than the GS treatment groups. This could be the result of the brain injury, which in the GS-treated groups is less than the control group, due to the reduction in plasma ammonia levels. The greater expression of neuroprotective genes in the control group corresponds to the greater severity of the brain injury, which is lower in the GS-treated groups. Overall, we have demonstrated successful reduction of ammonia in vivo using DNA-encoded GS enzyme therapy. This platform cannot compete with hemodialysis, however, could be used for cases with plasma ammonia levels below the critical 500 μM threshold levels. Additionally, there is a time delay for DNA-encoded enzyme expression to reach efficient enzyme expression for therapeutic efficacy, which hinders its use in acute disease applications with overly elevated ammonia levels. With regard to delivery and plasmid dosage of DNA-encoded enzymes for translatability to larger animals and humans, a study by Esquivel et al. [40] in nonhuman primates using DNA-encoded monoclonal antibodies showed administration of higher plasmid doses of up to 6 mg per site, and sequential administrations for a total of 18 mg total DNA, demonstrating significant therapeutic efficacy. Similar dosing regimens could be used to evaluate DNA-encoded enzyme therapy in humans. Considering large amount of skeletal muscle sites available for electroporation during each treatment session, administration of higher doses would be possible. This initial study provides important data on this approach as a new method to treat hyperammonemia. Such DNA-encoded, enzyme-based therapeutics could provide a complementary or alternative, potent, and cost-effective approach to reducing ammonia levels in hyperammonemic patients. Further study of this approach is warranted.
Footnotes
Acknowledgment
The authors thank The Wistar Genomics Core for performing the brain tissue RNA extraction and mRNA profiling using the NanoString nCounter Analysis System.
Author Disclosure Statement
L.H. is employee of Inovio Pharmaceuticals and as such receive salary and benefits, including ownership of stock and stock options. D.B.W. discloses grant funding, industry collaborations, speaking honoraria, and fees or stock for consulting. His service includes serving on scientific review committees and advisory boards. Remuneration includes direct payments and/or stock or stock options. K.M. reports receiving grants from Inovio, and receiving consulting fees from Inovio related to DNA vaccine development. The other authors declare no competing financial interests.
Funding Information
This work was supported by funding from Inovio Pharmaceuticals.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.