
Abstract
Type 2 diabetes (T2D) is a chronic metabolic disorder characterized by persistent hyperglycemia resulting from inefficient signaling and insufficient production of insulin. Conventional management of T2D has largely relied on small molecule-based oral hypoglycemic medicines, which do not halt the progression of the disease due to limited efficacy and induce adverse effects as well. To this end, antisense oligonucleotide has attracted immense attention in developing antidiabetic agents because of their ability to downregulate the expression of disease-causing genes at the RNA and protein level. To date, seven antisense agents have been approved by the United States Food and Drug Administration for therapies of a variety of human maladies, including genetic disorders. Herein, we provide a comprehensive review of antisense molecules developed for suppressing the causative genes believed to be responsible for insulin resistance and hyperglycemia toward preventing and treating T2D.
Introduction
Antisense oligonucleotides (AOs) are a class of synthetic nucleic acid molecules that are capable of regulating gene expression through binding to premature or mature messenger RNA (mRNA) targets [1–4]. Since the publication of seminal articles reporting AO-mediated inhibition of gene expression by Paterson et al., Zamecnik et al., and Stephenson et al. in the late 1970s [5–7], the therapeutic potential of AOs has been extensively explored.
To date, seven AOs have been granted approval by the United States Food and Drug Administration (FDA) as therapies for different diseases, as shown in Table 1 [8–29]. Approvals of these AO drugs have paved the way for researchers across academic and industry to develop AO-based therapeutics targeting different human maladies by either knocking down the expression of disease-causing genes or rescuing/upregulating the expression of essential or defective genes.
Antisense Oligonucleotides Approved by the United States Food and Drug Administration for Clinical Applications
AO, antisense oligonucleotide; CMV, cytomegalovirus; DMD, Duchenne muscular dystrophy; SMA, spinal muscular atrophy.
Milasen is a customised oligonucleotide drug for one person.
Diabetes is a chronic disorder that impairs body's ability to metabolize glucose, thus causing persistent hyperglycemia. According to International Diabetes Federation, more than 415 million adults had diabetes in 2015, and this number is expected to reach 642 million by 2040 [30]. By clinical presentation and etiology, diabetes can be broadly classified into two types: type 1 diabetes (T1D) and type 2 diabetes (T2D).
T1D, accounts for 5%–10% of cases, is characterized by autoimmune destruction of insulin-producing pancreatic β cells leading to absolute deficiency of insulin [31]. In contrast, T2D accounts for ∼90% of all cases and is characterized by inefficient signaling of insulin (leading to diminished response to insulin, usually referred to as insulin resistance or insulin insensitivity) in glucose recipient organs and tissues (muscle, adipose, liver, and brain), and insufficient production of insulin from β cells [32–35]. Insulin resistance exists before the onset of T2D [36]. Insulin is ineffective during insulin resistance state, and this is initially compensated by increased production of insulin to maintain glucose homeostasis, but over time, insulin production fails and insulin secretion decreases due to β cell dysfunction, resulting in T2D [31].
Obesity is closely associated with the initiation of insulin resistance and T2D [35]. Most of the T2D patients are obese (with higher body fat percentage). In obese individuals, enlarged adipose cells are resistant to the insulin-mediated anti-lipolytic effect and their capacity to store fat is diminished, resulting in increased free fatty acid (FFA) level in plasma [37]. Chronically elevated FFA reaches toxic levels and causes deleterious effects (known as lipotoxicity) within non-adipose tissues [37]. Lipotoxicity is a major cause of both insulin resistance and β cell dysfunction [38]. Therefore, obesity increases the risk for T2D through inducing insulin resistance [39,40].
As the level of circulating FFA is elevated in obese individuals [31,41], cellular uptake of FFA by muscle and liver is in excess [42–44]. Consequently, FFA triggers sequential activation of protein kinase C (PKC), IκB-kinase (IκK), and nuclear factor κB (NFκB) that stimulates the release of inflammatory chemokines, such as tumor necrosis factor-α (TNFα) and interleukin 6 (IL6), which further stimulate Jun N-terminal kinase-1 (JNK1) and suppress cytokine signaling-3 (SOCS3), respectively (Fig. 1) [45–47]. Collectively, JNK1 and SOCS3 downregulate the proteins participating in the insulin signal transduction pathway, including insulin receptor substrate (IRS), phosphatidylinositol 3-kinase (PI3K), and protein kinase B (Akt), thus preventing the translocation of glucose transporter type 4 (GLUT4) from the cytoplasm to cell membrane, which eventually inhibits the cellular uptake of glucose, leading to hyperglycemia (Fig. 1) [33,47].
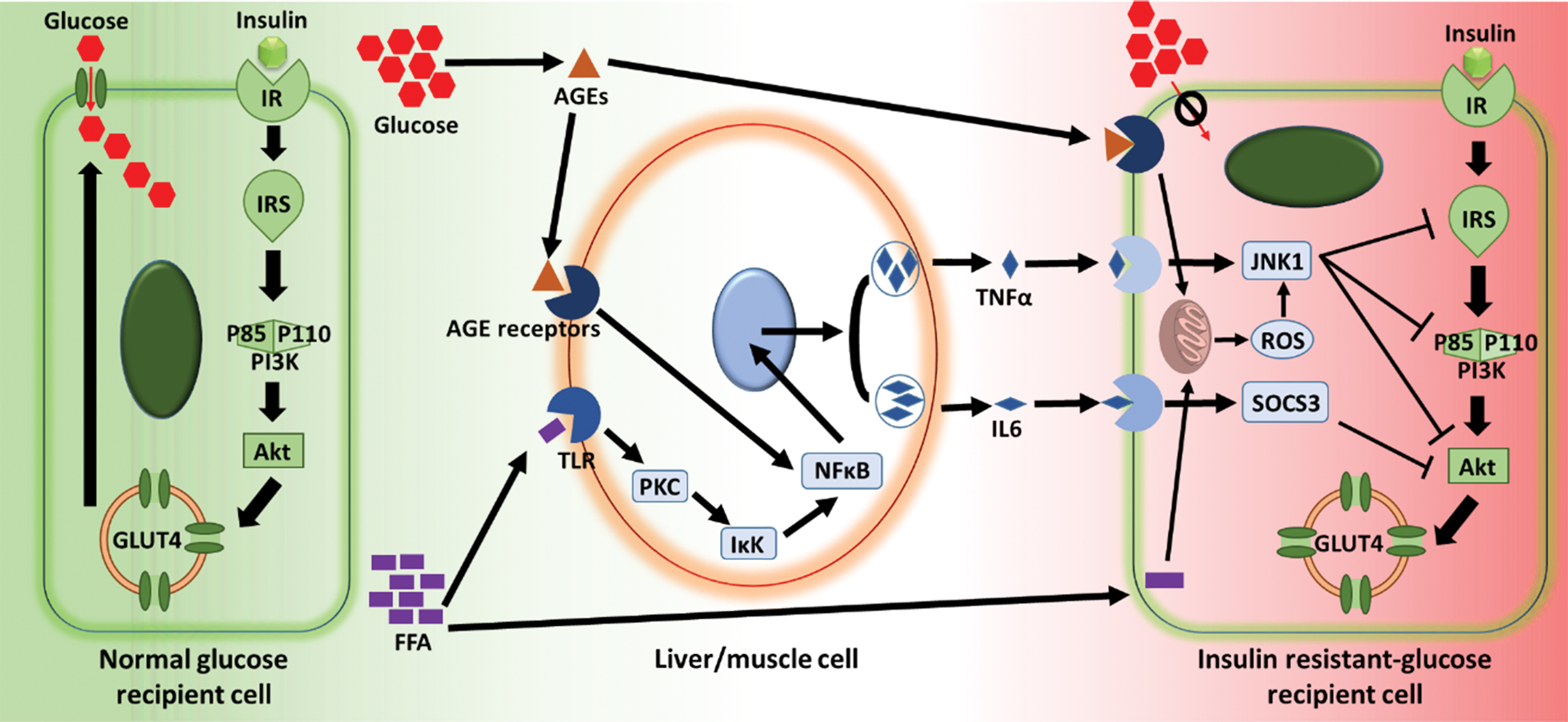
Mechanism of IR. IR, insulin receptor; IRS, insulin receptor substrate; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; GLUT4, glucose transporter type 4; FFA, free fatty acid; AGE, advanced glycation end product; PKC, protein kinase C; IκK, IκB-kinase; NFκB, nuclear factor κB; TNFα, tumor necrosis factor-α; IL6, interleukin 6; JNK1, Jun N-terminal kinase-1; ROS, reactive oxygen species; SOCS3, suppressor of cytokine signaling-3; glucose recipient cell represents cells from glucose recipient organs/tissues, including muscle, fat, liver, and brain. Color images are available online.
On the other hand, FFAs enter the mitochondria and participate in the tricarboxylic acid cycle, resulting in the generation of reactive oxygen species (ROS) that also triggers JNK1 (Fig. 1) [48]. Furthermore, in chronic state of hyperglycemia, glucose also interacts with proteins through nonenzymatic glycation and oxidation processes, leading to the formation of advanced glycation end products that in turn aggravate insulin resistance through triggering ROS generation and NFκB (Fig. 1) [49]. Besides, glucotoxicity, lipotoxicity, and inflammation caused by hyperglycemia and high FFA concentration impair the pancreatic β cells and finally exhaust their capability to secrete insulin, as a result of cellular dysfunction and apoptosis [50–53].
Currently prescribed small molecule-based antidiabetic drugs mainly aim at reducing blood glucose level by targeting key glycometabolic organs, including the small intestine, pancreas, muscle, liver, fat, and kidney [54]. So far, several classes of oral hypoglycemic drugs have been used such as glitazones, meglitinides, biguanides, sulfonylureas, α-glucosidase inhibitors, sodium glucose cotransporter-2 (SGLT2) inhibitors, dipeptidyl peptidase-4 inhibitors, and glucagon-like peptide-1 (GLP1) receptor agonists [55–57]. However, these agents do not halt the progression of T2D due to two reasons: first, few of the agents target the underlying root cause of the disease: insulin resistance; and second, as small-molecule drugs, they do not interfere with the transcription or translation processes of the disease-causing genes, and thus show lack of efficacy compared to RNA-targeting nucleic acid interventions, for example, AO and small interfering RNA (siRNA). In addition, oral hypoglycemic drugs induce adverse reactions, affecting patient compliance.
Recently, Kokil et al. comprehensively reviewed the mechanisms and limitations of conventional hypoglycemic agents and elaborated the potential of siRNA as a novel therapy for T2D [33]. Herein, we outline another option of RNA therapeutics: AO interventions to modify expression of genes implicated in the T2D pathogenesis, particularly, insulin resistance.
AOs As Therapeutics: Mechanisms of Action
AOs are single-stranded nucleic acids that contain 15–30 nucleotides. AOs can be used to manipulate target gene expression through specifically binding to target RNAs by Watson–Crick base pairing [58]. Basically, there exists three distinct mechanisms of action: (1) induction of RNase H-mediated degradation of mRNA; (2) modulation of splicing by sterically blocking the binding of splicing factors to the precursor mRNA (pre-mRNA); and (3) translational repression by either sterically blocking the 5′ untranslated region in pre-mRNA to avoid 5′ cap formation or hindering the association of translation machinery to mRNA (Fig. 2) [2,59–63]. To date, four mRNAs degrading AOs and four splice modulating AOs have been approved for clinical use.

Schematic illustration of the mechanisms underlying AO-mediated regulation of gene expression.
RNase H recruitment to form an AO:mRNA duplex and the subsequent hydrolysis of mRNA is the most widely utilized mechanism for potent downregulation of target gene expression. To date, four AOs evoking this mechanism have reached the market: fomivirsen (VitraveneTM), mipomersen (KynamroTM), inotersen (TegsediTM), and volanesorsen (WaylivraTM). Their specific mechanisms are described below:
Fomivirsen (Vitravene) binds to and degrades the mRNAs encoding cytomegalovirus (CMV) immediate-early 2 protein (required for viral replication), thus providing therapeutic effects for CMV retinitis by inhibition of virus proliferation [8,9]. Mipomersen (Kynamro) binds to and degrades the mRNAs encoding apolipoprotein B [a component of low-density lipoprotein cholesterol (LDL-C)], thus providing therapeutic effects for familial hypercholesterolemia through reducing atherogenic LDL-C production [10,11]. Inotersen (Tegsedi) binds to and degrades the mRNAs encoding transthyretin (TTR), thus providing therapeutic effects for hereditary TTR amyloidosis by knocking down the expression of mutant TTR that causes amyloid aggregates to deposit in organs [24,25]. Volanesorsen (Waylivra) binds to and degrades the mRNAs encoding apolipoprotein CIII, a key component in hypertriglyceridemia and chylomicronemia, thus providing therapeutic effects for familial chylomicronemia syndrome through promoting triglyceride clearance and decreasing plasma triglyceride levels [64].
Splice modulation is mainly applied to remove or prevent premature termination codons (PTCs), caused by loss-of-function mutations, through exon skipping or exon inclusion, in an attempt to restore the open reading frame (ORF) and rescue or increase the production of functional essential proteins [60]. Alternatively, AO-mediated exon skipping can also be used to induce a frameshift and elicit PTCs that lead to knockdown of target gene expression [65].
Four splice-modulating AOs have been approved by FDA for the treatment of genetic diseases: eteplirsen (Exondys 51TM), golodirsen (Vyondys 53TM), nusinersen (SpinrazaTM), and milasen. Their specific mechanisms are described below:
Eteplirsen (Exondys 51) and golodirsen (Vyondys 53) induce the skipping of exon 51 and 53 respectively, in the dystrophin mRNA. This overcomes PTCs resulting from frameshift deletions and in turn restores the dystrophin ORF and allows the synthesis of truncated, but partially functional dystrophin proteins. This is highly beneficial for patients with Duchenne muscular dystrophy (DMD) [12–15,29].
Nusinersen (Spinraza) retains exon 7 in the mRNA of survival of motor neuron 2 (SMN2), preventing loss of function resulting from natural skipping of exon 7, and increasing the production of full-length, functional SMN protein that has shown clinical benefit in patients with spinal muscular atrophy [16–23].
Milasen skips a transposon-activated cryptic exon located between exon 6 and 7 in major facilitator superfamily domain containing 8 (MFSD8) mRNA, thereby evading the PTC in the cryptic exon, and rescuing functional MFSD8 synthesis, providing a molecular therapy for ceroid lipofuscinosis type 7 related Batten disease [26–28].
Chemical Modifications and Designs of AOs
AOs composed of natural nucleotides, that is, ribonucleotides or deoxyribonucleotides, are not suitable for drug development applications due to several limitations such as lack of resistance to nucleases, poor binding specificity, and affinity to target RNAs [66]. To improve the drug-like properties of AOs, different types of chemically modified nucleotides are introduced into AOs during synthesis (Fig. 3) [66–71].
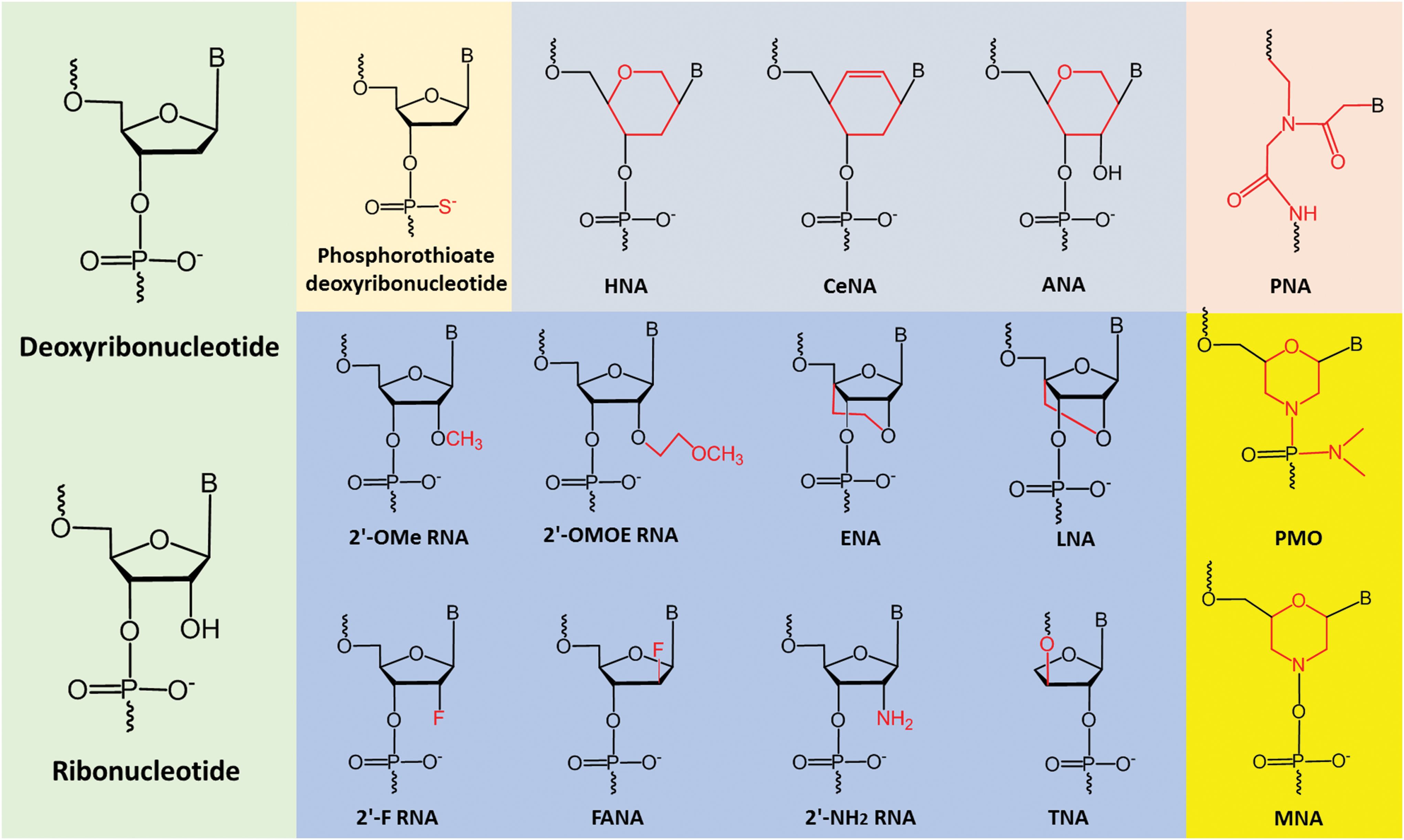
Examples of nucleotide analogs. 2′-OMe, 2′-O-methyl; 2′-OMOE, 2′-O-methoxyethyl; ENA, 2′-O, 4′-C-ethylene-bridged nucleic acid; LNA, locked nucleic acid; 2′-F, 2′-fluoro; 2′-FANA, 2′-fluroarabino nucleic acid; 2′-NH2, 2′-amino; TNA, threose nucleic acid; HNA, 1,5-anhydro hexitol nucleic acid; CeNA, cyclohexenyl nucleic acid; ANA, altritol nucleic acid; PNA, peptide nucleic acid; PMO, phosphorodiamidate morpholino oligomer; MNA, morpholino nucleic acid. Color images are available online.
The first generation of modification includes oligonucleotides with modified internucleotide phosphate linkages, called phosphorothioate (PS) in which the nonbridging oxygen atom within phosphate group is replaced by a sulfur atom (Fig. 3) [72]. PS backbone confers higher nuclease resistance and prolonged half-life in plasma. The second generation of modification contains a series of nucleotides with 2′-modified sugar moiety. The most widely used analogs of this class are 2′-O-methyl (2′-OMe), 2′-Fluoro (2′-F), and 2′-O-methoxyethyl (2′-OMOE) (Fig. 3). AOs with such modifications possess improved nuclease resistance and higher target binding affinity [73–75].
Third generation of modified nucleotides is usually with structural changes of larger extent compared to the first and second generations. For example, locked nucleic acid (LNA) has an extra bridge connecting the 2′ oxygen and 4′ carbon in the sugar moiety (Fig. 3) [76,77]; moreover, the backbones of phosphorodiamidate morpholino oligomer (PMO) and peptide nucleic acid (PNA) are even completely different from natural DNA/RNA. PMO contains a morpholine ring with a phosphorodiamidate linkage group instead of deoxyribose-/ribose-phosphate structure, and PNA's backbone is composed of N-(2-aminoethyl)-glycine with peptide bonds (Fig. 3) [78–82]. Analogs of this generation exhibit excellent nuclease stability and hybridization affinity.
Although uniformly modified 2′-OMe, 2′-OMOE, or PMO AOs are resistant to nuclease and form more stable duplex with their target RNA than the AOs constructed by deoxyribonucleotides, they are not substrates of RNase H. One solution is the design of gapmer AO (Fig. 4) in which a central DNA region containing ∼10 deoxyribonucleotides (with or without PS linkages) sufficient to recruit RNase H is flanked by sequences consisting of around 5–7 modified nucleotides, including 2′-OMe or 2′-OMOE analogs at each end [83–85]. Gapmer-like AO balances the capability of inducing RNase H-mediated target RNA degradation and the protection of AO from nuclease attacks.

Schematic illustration of AOs with uniformly modified analogs and AOs composed of nucleotides of different chemistries (chimeric AO: gapmer and mixmer). Color images are available online.
However, PMO is not compatible with other chemistries to synthesize chimeric AOs; as a result, PMO AOs are solely utilized for splice modulation or translational repression. Interestingly, Veedu and colleagues recently developed an analog of PMO called morpholino nucleic acid to synthesize chimeric AOs in combination with 2′-OMe-PS analogs [86], enabling nucleotide analog with morpholine ring to associate with other chemistries to form gapmers or mixmers. In fact, mixmer design (Fig. 4) has been widely investigated in optimizing the efficacy of splice-modulating AOs other than RNase H-inducing AOs [87–91].
It is noteworthy mentioning that PS, 2′-OMOE, and PMO are the only three chemistries applied in the FDA-approved AO drugs: fomivirsen (Vitravene) contains 21 deoxyribonucleotides on a PS backbone [8,9], both mipomersen (Kynamro) and inotersen (Tegsedi) are of 5–10–5 gapmers on a PS backbone in which the middle ten deoxyribonucleotides are flanked by five 2′-OMOE analogs at both ends [10,11,24,25], nusinersen (Spinraza) and milasen are uniformly modified 2′-OMOE AOs with PS linkages of 18 and 22 nucleotides, respectively [16–23,26–28], and eteplirsen (Exondys 51) and golodirsen (Vyondys 53) are PMO AOs with 30 and 25 monomers, respectively [12–15,29].
AO Delivery
Delivery of AO drugs still remains one of the major challenges. Different methods have been developed and explored in assisting AOs to deliver into target cells, for example, liposomes, cell-penetrating peptides, N-acetylgalactosamine (GalNAc), antibodies, and aptamers [92–100]. Recently published reviews on this topic have comprehensively described and discussed different delivery methods and mechanisms of nucleic acid-based therapeutics in the aspects of cellular uptake, biodistribution, and pharmacokinetics [101–105].
However, it is noteworthy mentioning that so far, the FDA-approved AO drugs are directly administered without any assistance of the abovementioned delivery agents: fomivirsen (Vitravene) is delivered through intraocular injection [8,9], both mipomersen (Kynamro) and inotersen (Tegsedi) are administered by subcutaneous injection (SCI) [10,11,24,25], nusinersen (Spinraza) and milasen are introduced through intrathecal injection [16–23,26–28], and intravenous infusion (IVI) is used for eteplirsen (Exondys 51) and golodirsen (Vyondys 53) [12–15,29].
AOs preferentially accumulate in liver and kidney when delivered systemically [106]. IVI or intraperitoneal injection (IPI) of AOs hardly enters the brain due to physiological obstruction caused by blood–brain barrier [107,108]. This problem can be solved by intracranial delivery, including local injections of AOs in the desired brain region or AO injection in the cerebrospinal fluid [109]. Infusion of AOs into primate brains showed AO dispersion throughout the brain, spinal cord, and brain stem [109].
Apart from selective injection approach (eg, intracranial delivery), chemical modification also confers AO improved cellular delivery. The protein-binding properties of PS modification facilitate interactions of PS-AOs with acceptor proteins on the cell membrane, leading to endocytosis-mediated internalization of PS-AOs into target cells [110]. For instance, PS-AOs interact with asialoglycoprotein receptor (ASGR), which is highly expressed by hepatocytes [111], thereby improving their uptake of PS-AOs in liver [112].
Furthermore, cell-specific delivery can be achieved by using ligand-receptor systems. For example, GalNAc/ASGR is a natural ligand/receptor system. GalNAc conjugation to AOs significantly enhances AO uptake by hepatocytes [110]. Another example is GLP1 conjugation, which significantly enhances the uptake of GLP1-conjugated AOs by pancreatic β-cells (β-cells are among the cells refractory to AO uptake) through interactions of GLP1 and its receptor, GLP1R, on β-cell surface [106,110].
AOs Targeting T2D
Compared to the steric blocking AOs that either modulate splicing or repress translation, gapmer-like AOs that knock down gene expression through RNase H mechanisms are predominantly utilized in the modification of target gene expression involved in T2D pathogenesis and the development of AO-based therapeutics. In this section, we focus on chemically modified AOs that impede production of proteins involved in insulin resistance and hyperglycemia.
These include protein tyrosine phosphatase-1B (PTP1B, encoded by PTPN1), glucagon receptor (GCGR, encoded by GCGR), SGLT2 (encoded by SLC5A2), glucocorticoid receptor (GCCR, encoded by NR3C1), acetyl-CoA carboxylase 1 or 2 (ACC1 or ACC2, encoded by ACACA or ACACB), diacylglycerol acyltransferase-2 (DGAT2, encoded by DGAT2), mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M, encoded by PCK2), protein kinase C-ɛ (PKCɛ, encoded by PRKCE), glucose 6-phosphate transporter-1 (G6PT1, encoded by SLC37A4), cAMP response element binding protein (CREB, encoded by CREB), CREB-regulated transcription coactivator-2 (CRTC2, encoded by CRTC2), forkhead box O1 (FOXO1, encoded by FOXO1), density-enhanced phosphatase-1 (DEP1, encoded by PTPRJ), retinol binding protein 4 (RBP4, encoded by RBP4), TTR (encoded by TTR), tribbles homolog 3 (TRIB3, encoded by TRIB3), IκB-kinase β (IκKβ, encoded by IKBKB), and toll-like receptor 2 (TLR2, encoded by TLR2).
Protein tyrosine phosphatase-1B
PTP1B negatively regulates insulin signaling by dephosphorylating the upstream proteins in the insulin signal transduction pathway, including insulin receptor (IR) and IRS [113,114]. The AO ISIS 113715, developed by Ionis Pharmaceuticals, is a 5–10–5 2′-OMOE gapmer AO on PS backbone that downregulates the expression of PTP1B protein [115]. ISIS 113715 is utilized in the characterization of PTP1B function, since the therapeutic potential of this AO for T2D has been investigated.
Zinker et al. reported that IPI of ISIS 113715 into ob/ob and db/db mice resulted in significant reduction of PTP1B expression in both liver (ob/ob mice treated by 25 mg/kg for 6 weeks: −50% and db/db mice treated by 50 mg/kg for 4 weeks: −50%) and fat (ob/ob mice treated by 25 mg/kg for 6 weeks: −60%), with reduced hyperinsulinemia (ob/ob mice treated by 25 mg/kg for 6 weeks: −77%) and increased expression of proteins involved in the insulin signaling pathway, including IRS2, PI3K p50α, and p55α isoforms [115]. On the other hand, the expression of p85α was decreased [115]. It is noteworthy that increased expression of p50α and p55α isoforms instead of p85α was reported to improve insulin sensitivity [116]. Besides, AO treatment also led to reduced expression of enzymes related to gluconeogenesis such as PEPCK and fructose-1,6-bisphosphatase (FBP) [115].
Collectively, changes in protein expression levels as a result of ISIS 113715 treatment correlated with increased Akt phosphorylation in liver, improved insulin sensitivity, and normalization of hyperglycemia as plasma glucose, postprandial glucose excursion, and hemoglobin A1C were corrected to normal levels [115].
Rondinone et al. provided more evidence on the differential expression of PI3K isoforms induced by PTP1B reduction [117]. ob/ob mice dosed with 50 mg/kg ISIS 113715 for 3 weeks exhibited downregulation of p85α isoform and upregulation of p50α and p55α isoforms in both liver and fat [117]. Mouse hepatocytes treated with an AO targeting p85α (ISIS 131410) exhibited reduction in p85α expression and increased production of p50α and p55α that correlated with enhanced Akt phosphorylation, indicating an increase in insulin sensitivity [117].
Apart from modulating the expression levels of proteins involved in the insulin signaling pathway, Clampit et al. found that PTP1B reduction directly leads to improved insulin sensitivity by modulating key signal transduction events: tyrosine phosphorylation of those proteins [118]. In that in vitro study, up to 70% decrease of PTP1B expression in FAO rat hepatoma cells was achieved by transfecting the cells with ISIS 113715 at a very high level (20 μM) through electroporation. Subsequent insulin stimulation resulted in elevated tyrosine phosphorylation of IR, IRS, Akt, and glycogen synthase kinase 3 (GSK3, substrate of Akt), indicating an ameliorated insulin signaling efficiency [118].
A report from Gum et al. illustrated that the reduction of PTP1B is sufficient to improve insulin sensitivity, at least in an animal model [119]. In their study, ob/ob mice were treated with ISIS 113715 at a dosage of 25 mg/kg for 6 weeks leading to 60% decrease of PTP1B protein in liver. Subsequent insulin stimulation resulted in elevated tyrosine phosphorylation of IR, IRS1, IRS2, PI3K, Akt, and GSK3 by threefold, fourfold, threefold, threefold, sevenfold, and more than twofold, respectively. Although the PTP1B level was not reduced in muscle, increased Akt phosphorylation was observed, demonstrating that insulin resistance is ameliorated by PTP1B targeting AO treatment in vital peripheral tissues [119].
Rondinone et al. demonstrated that PTP1B plays an important role in lipogenesis, fat storage, and the development of obesity [120], and thus contributes to insulin resistance and T2D. In their study, administration of ISIS 113715 into ob/ob mice at a dosage of 25 mg/kg for 6 weeks decreased the level of a number of proteins involved in lipogenesis, such as sterol regulatory element-binding protein 1, fatty acid synthase (FAS), and spot14. Reduction of these proteins correlated with significant decrease of fat depot (−42%) and triglyceride content (−25%) in adipocytes compared to the control group (saline treatment) [120]. In addition, AO treatment increased IRS2 and PI3K p50α isoforms, and also decreased p85α in fat [120], suggesting that insulin signal transduction was improved.
Similarly, Waring et al. also studied the effect of PTP1B reduction on the expression of lipogenesis-related genes by treating ob/ob mice with ISIS 113715 for 6 weeks [121]. They found that PTP1B reduction not only led to downregulation of lipogenetic genes in fat but also in liver [121]. In addition, the expression of genes involved in adipocyte differentiation such as malic enzyme, adipsin, and retinol-binding protein was also downregulated as a result of PTP1B inhibition [121]. These findings provide further evidence that PTP1B plays an important role in enlargement of fat storage and the development of obesity.
Swarbrick et al. reported that the improvements of hyperglycemia and hyperinsulinemia resulting from PTP1B reduction correlated with the increase of adiponectin [122], a fat-derived hormone that strengthens insulin signaling mainly by activating IRS [123]. In a non-human primate study conducted by this group, obese, insulin-resistant rhesus monkeys were treated by ISIS 113715 with a dosage of 20 mg/kg for 4 weeks. Fasting concentrations of plasma glucose and insulin were reduced, insulin sensitivity was increased, and the monkeys receiving 4 weeks of treatment exhibited marked increase of adiponectin concentration by 70%, which was in proportion to the amelioration of insulin sensitivity [122].
Gum et al. discovered that ameliorated hyperglycemia and hyperinsulinemia resulting from PTP1B reduction correlate with inhibition of p38 mitogen-activated protein kinase (p38 MAPK) phosphorylation and the subsequent inhibition of its substrates: TNFα and CREB [124]. TNFα is associated with insulin resistance [37,125,126], and CREB contributes to hyperglycemia by promoting gluconeogenesis through induction of PEPCK transcription [127,128]. In this study, ob/ob mice treated by 25 mg/kg of ISIS 113715 for 6 weeks led to significantly decreased phosphorylation of p38 MAPK and CREB by 70% and 80%, respectively, in contrast to the mice treated with saline. ISIS 113715-treated mice also showed ∼37% of reduction in TNFα expression [124].
In an attempt to optimize the efficacy of ISIS 113715, Koizumi et al. synthesized a 5–10–5 ethylene-bridged nucleic acid (ENA) gapmer named ENA-1 (chemical structure of ENA is shown in Fig. 3) with the identical nucleobase sequence to ISIS 113715 and compared the antidiabetic effect of these two AOs [129]. Their results showed that PTP1B expression in db/db mice was reduced more effectively when treated with ENA-1 than ISIS 113715 at a dosage of 60 mg/kg, either through SCI or IPI (they tried 45 mg/kg IPI at first, but the difference of activity between two AOs was not apparent, so they shifted to 60 mg/kg for better comparison).
Furthermore, plasma glucose was decreased to a larger extent in ENA-1-treated mice compared to those treated by ISIS 113715 [129]. In addition, the melting temperature (Tm) of ENA-1 (77°C) was higher than ISIS 113715 (62.5°C), indicating that ENA-1 possesses higher binding affinity than ISIS 113715. The distribution of ENA-1 in both liver and fat, 72 h after administration, was also found to be at least twofold higher compared with ISIS 113715, which was probably due to the higher nuclease stability of ENA compared to the 2′-OMOE chemistry. Altogether, these results suggest that ENA modification may be a better option than 2′-OMOE in constructing gapmer AOs targeting PTP1B [129].
As the therapeutic effect of PTP1B reduction on obesity-associated T2D has been widely explored, Pandey et al. investigated the role of low molecular weight PTP (LMW-PTP) in T2D through downregulating LMW-PTP expression by a specific AO, ISIS 288267 in diet-induced obese (DIO) mice and ob/ob mice [130]. AO-induced LMW-PTP reduction led to increased phosphorylation of IR, PI3K, and Akt in mice treated with 25 mg/kg of ISIS 288267 for 4 weeks (twice a week). In addition, ∼42% and 20% decreases of fed plasma insulin and glucose in DIO mice, and ∼47% and 42% decreases of fasted plasma insulin and glucose in ob/ob mice were observed, respectively, indicating an increased insulin sensitivity resulting from LMW-PTP reduction [130]. This research has demonstrated a clear similarity between LMW-PTP and PTP1B in terms of insulin resistance. Therefore, LMW-PTP is a potential target for T2D therapy.
Glucagon receptor
Glucagon is a hormone produced by pancreatic α cells that stimulates glucose production through two pathways: glycogenolysis mediated by cAMP-related signaling and gluconeogenesis through activation of multiple enzymes [131,132]. Excess glucagon leads to hyperglycemia [133].
Liang et al. developed a 5–10–5 2′-OMOE-PS gapmer AO (ISIS 148359) that reduces the expression of GCGR in an attempt to block glucagon signal transduction and thus ameliorate hyperglycemia and T2D [134]. In this study, db/db mice treated with 25 mg/kg of ISIS 148359 through IPI for 3 weeks (twice a week) resulted in decreased GCGR expression and hepatic cAMP production stimulated by glucagon. This correlated with significant decreases of serum glucose (−24%), triglyceride (−62%), and FFA (−36%) [134]. Hypoglycemia was not identified in the mice receiving AO treatment, in support of the concept that GCGR could be a therapeutic target in controlling hyperglycemia caused by T2D [134].
To further characterize the mechanisms underlying amelioration of T2D mediated by GCGR reduction, Sloop et al. treated rodent models with GCGR targeting AOs (ISIS 148359 and ISIS 180475), leading to decreased production of enzymes related to gluconeogenesis and glycogenolysis, normalized plasma glucose, increased GLP1 production (GLP1 promotes insulin secretion in a glucose-dependent manner [135]), and preserved insulin secretion [136].
Zucker diabetic fatty (ZDF) rats treated with 25 mg/kg ISIS 180475 for 4.5 weeks (SCI twice a week) exhibited reduced gluconeogenetic or glycogenetic enzyme levels, such as glucose-6-phosphatase catalytic subunit (G6P [cat]), PEPCK cytosolic isoform (PEPCK-C), and glycogen phosphorylase reduction by about 65%, 78%, and 45%, respectively, and decreased plasma glucose by ∼66% compared to the saline control group. The amount of islet GLP1 and insulin in db/db mice treated by ISIS 180475 was 3.5- and 2-fold higher than in the saline-treated control group, respectively [136]. This study demonstrated that attenuation of glucagon action resulting from GCGR reduction improves hyperglycemia through decreasing glucose production and ameliorating pancreatic β cell function.
Sodium glucose cotransporter-2
SGLT2 plays a vital role in renal glucose reabsorption. SGLT2 inhibition increases urinary glucose excretion, decreases blood glucose levels, and thus presents a therapeutic option for hyperglycemia and T2D [137].
Zanardi et al. treated mice and cynomolgus monkeys with an SGLT2 targeting short 2–8–2 2′-OMOE-PS gapmer AO (ISIS 388626) at various SCI doses (1, 3, 10, and 30 mg/kg/week), and achieved decreased renal SGLT2 mRNA production, in a dose-dependent manner, after 13 weeks of treatment (mice: 18%, 5%, 4%, and 2% and monkey: 64%, 53%, 31%, and 10%) compared to the saline controls [138]. Reduction of SGLT2 expression correlated with increased urine glucose elimination, although dose-dependent basophilic granule accumulation was identified in renal tubular epithelial cells due to AO accumulation, but no functional kidney change was observed [138]. These results indicated that ISIS 388626 may be an effective SGLT2 inhibitor with a satisfactory safety profile, and further investigation in clinical trials was recommended [138].
Glucocorticoid receptor
Glucocorticoids are steroid hormones that regulate glucose homeostasis. Glucocorticoids promote gluconeogenesis in liver [139,140], reduce glucose uptake in muscle and fat [141,142], and inhibit glycogen synthesis in skeletal muscle [143,144] by antagonizing insulin response. Overall, these effects are important for metabolic adaptation during stress, such as starvation, when blood glucose needs to be preserved for brain, as transiently raising blood glucose is critical to promote maximal brain function [145]. However, excess glucocorticoids contribute to hyperglycemia [146]. Inhibition of the action of GCCR could be a therapeutic approach for T2D.
Watts et al. treated different rodents [ob/ob mice, db/db mice, ZDF rats, and high-fat diet (HFD)-fed rats] with optimized GCCR targeting 2′-OMOE-PS gapmer AOs for 4 weeks (up to 25 mg/kg, SCI) resulting in significant decreases of GCCR expression in both liver and fat, which were accompanied by ∼30%, 40%, 40%, and 70% decreases in fasted glucose levels in ob/ob mice, db/db mice, ZDF rats, and HFD-fed rats, respectively [147]. Furthermore, a 60% decrease in insulin levels and 35% decrease of TNFα were observed in ob/ob mice, and a reduction of plasma triglycerides was observed in both db/db mice and ZDF rats [147].
This research indicated that glucocorticoid antagonism through AO-mediated GCCR inhibition is a promising antidiabetic approach. At the same time, Liang et al. provided similar evidence that GCCR inhibition leads to alleviation of hyperglycemia in ob/ob and db/db mice through IPI administration of a mouse-specific, GCCR targeting 5–10–5 2′-OMOE-PS gapmer AO (ISIS 180272) at a dosage of 25 mg/kg for 3 weeks [148]. AO treatment lowered plasma glucose levels by 45% and 23% in ob/ob and db/db mice, respectively, and a marked reduction in the activity of gluconeogenetic proteins such as PEPCK and G6P was identified [148].
Acetyl-CoA carboxylase
Nonalcoholic fatty liver disease (NAFLD) caused by hepatic triglyceride accumulation is responsible for insulin resistance and elevated glucose production, which are key elements of T2D [149–151]. Therefore, reducing lipid accumulation in liver could be a promising strategy for T2D therapy. ACC catalyzes the synthesis of malonyl-CoA, a regulator of both synthesis and oxidation of fatty acid [152–155]. ACC has two isoforms: ACC1 and ACC2 [156].
To explore the impact of downregulating ACC1, ACC2, and both ACC1 and ACC2 on lipid levels and insulin sensitivity in liver, Savage et al. developed three 5–10–5 2′-OMOE-PS gapmer AOs targeting ACC1 (ISIS 338292), ACC2 (ISIS 189594), and both ACC1 and ACC2 (ISIS 362037), and treated rats with NAFLD with these AOs for ACC suppression [157]. Knockdown of either ACC1 or ACC2 with AOs promoted hepatic fat oxidation, while suppression of both ACC1 and ACC2 with ISIS 362037 increased fat oxidation to a significantly higher degree [157]. Interestingly, ACC1 reduction also resulted in inhibition of lipogenesis. Furthermore, ACC1/2 suppression by ISIS 362037 lowered the level of hepatic lipids such as diacylglycerol and triglycerides, and increased insulin sensitivity [157]. These results support ACC modulation as a novel approach to the treatment of insulin resistance and T2D.
Diacylglycerol acyltransferase-2
DGAT2 is an enzyme that participates in the synthesis of triglyceride [158]. In an attempt to investigate the role of DGAT2 in lipid metabolism, Yu et al. [158] treated both DIO mice and ob/ob mice with a DGAT2-specific, 5–10–5 2′-OMOE-PS gapmer AO (ISIS 217376) at a dose of 25 mg/kg through SCI. ISIS 217376 treatment decreased DGAT2 mRNA levels by more than 75% in liver and fat; this was accompanied by decreased triglyceride synthesis and increased fatty acid oxidation in mouse hepatocytes, and a marked reduction of triglyceride (−37%), FFA (−50%), and diacylglycerol (−74%) in liver compared to mice treated by control AO [158]. Reduction in mRNA levels encoded by lipogenic genes, for example, ACC1, ACC2, and FAS, were also observed [158], indicating that DGAT2 reduction negatively regulates lipogenesis in liver, thereby ameliorating hyperlipidemia and steatosis.
Later, Choi et al. [159] studied the antidiabetic effect of DGAT2 reduction by administering another DGAT2 targeting gapmer AO, ISIS 369235 (50 mg/kg, IPI), to rats with diet-induced NAFLD [135]. Similar to the report from Yu et al. [158], AO treatment led to significant reduction in hepatic diacylglycerol and triglyceride, and decreased expression of lipogenic genes. In addition, increased expression of genes related to lipid oxidation was also identified [159]. Importantly, changes of gene expression and lipid content level were accompanied with increased activity of PI3K and Akt, and decreased hepatic glucose production, indicating a significantly improved insulin sensitivity [159]. Therefore, AO-induced DGAT2 knockdown could be an antidiabetic strategy as it alleviates insulin resistance through lowering hepatic lipid content.
Mitochondrial phosphoenolpyruvate carboxykinase
Gluconeogenesis generates glucose from noncarbohydrate carbon substrates such as pyruvate, lactate, alanine, glutamine, and glycerol. Conversion of oxaloacetate to phosphoenolpyruvate is required for gluconeogenesis from pyruvate, which is catalyzed by PEPCK [160]. Generally, this reaction is attributed to the PEPCK-C [161]. Stark et al. hypothesized that PEPCK-M also contributes importantly to gluconeogenesis [162]. In this research, they silenced the expression of PEPCK-M in rats by administration of a PEPCK-M-specific, 5–10–5 2′-OMOE-PS gapmer AO (ISIS 421062) through IPI at a dosage of 25 mg/kg (twice weekly) for 4 weeks [162].
As a result, lowered plasma glucose, insulin, and triglycerides, and reduced white adipose tissue were observed. No hypoglycemia was identified and was accompanied with a switch of gluconeogenic substrate preference to glycerol, bypassing the PEPCK reaction [162]. Besides, normal mitochondrial function was maintained in PEPCK-M-deficient hepatocytes compared to PEPCK-C knockout that led to mitochondrial deficiency. This research pointed out the significance of the role of PEPCK-M in rodent gluconeogenesis, and suggests that perhaps PEPCK-M, instead of PEPCK-C, could be a therapeutic target for T2D in humans [162]; however, target verification is definitely required in future experiments.
Protein kinase C-ɛ
To investigate the role of PKCɛ in linking fat accumulation and insulin resistance in liver, Samuel et al. treated rats with HFD-induced hepatic insulin resistance by a PKCɛ-specific, 5–10–5 2′-OMOE-PS gapmer AO (ISIS 232697) with an IPI dosage of 75 mg/kg per week for 4 weeks [163].
It was found that PKCɛ, rather than other isoforms of PKC, was activated in insulin-resistant rats and PKCɛ knockdown by ISIS 232697 increased the hepatic insulin sensitivity [163]. The AO treatment led to markedly raised IRS2 tyrosine phosphorylation (∼300% above basal), which was accompanied by ∼350% increase of Akt2 activity when compared to the saline control. Significantly elevated Akt2 activity was also identified in adipose tissue in AO-treated rats (221%) compared to saline control (32%). Furthermore, it was found that PKCɛ impairs IR kinase activity both in vivo and in vitro [163]. Altogether, this research reveals the critical role of PKCɛ in fat-induced insulin resistance; therefore, PKCɛ inhibition could be a therapeutic strategy for NAFLD and T2D.
Glucose 6-phosphate transporter-1
G6P hydrolization and the subsequent glucose formation are catalyzed by G6P as the final step of gluconeogenesis; therefore, G6P plays a pivotal role in glucose homeostasis [164]. Sloop et al. hypothesized that inhibition of G6PT1 may provide a therapeutic effect on hyperglycemia and T2D. They treated ob/ob mice with a G6PT1 targeting 5–10–5 2′-OMOE-PS gapmer AO (ISIS 149008) with an SCI dosage of 25 mg/kg (twice a week) for 6 weeks [165].
The hepatic G6PT1 mRNA was decreased by 85% and the activity of G6P transport was reduced by 75%, compared to the control group. Inhibition of G6PT1 was correlated with blunted hepatic glucose production, reduced plasma glucose level, and improved hyperglycemia in mice [165]. In addition, hypoglycemia and other adverse metabolic effects were not identified [165]. According to these findings, AO-induced G6PT1 inhibition could lead to effective glucose control in T2D, without adverse effects on metabolism.
cAMP response element binding protein
CREB promotes glucose production by directly activating the transcription of key gluconeogenic genes such as PEPCK, G6P, and FBP due to the presence of a cAMP response element (CRE) at the promotor of these genes [166]. Erion et al. hypothesized that reduction of CREB expression could improve hyperglycemia resulting from insulin resistance [167].
In this study, rats with HFD induced-insulin resistance were treated by a CREB-specific 2′-OMOE-PS gapmer AO that improved insulin sensitivity. This was seen in a series of changes: 43%, 55%, and 54% reductions in the mRNA expression of the gluconeogenic proteins, PEPCK-C, PEPCK-M, and PGC1α, respectively; suppression of glucose production in liver by 45%; fasting plasma insulin level was reduced by 66%; hepatic and plasma triglycerides were decreased by 57% and 24%, respectively; and diacylglycerols in liver were lowered, accompanied by reduced PKCɛ membrane translocation [167]. This study demonstrates that AO-induced CREB reduction prevents hepatic steatosis and insulin resistance, and therefore, CREB is an attractive therapeutic target for T2D.
CREB-regulated transcription coactivator-2
CRTC2 is a coactivator of CREB that regulates transcription of gluconeogenic genes by enhancing binding of CREB to the target CRE within the specified cAMP-responsive genes [168,169]. Erion et al. investigated the effect of CRTC2 reduction on the control of glucose homeostasis by treating rats with T2D with a CRTC2 targeting, 5–10–5 2′-OMOE-PS gapmer AO (ISIS 384680) at an IPI dose of 37.5 mg/kg (twice a week) for 4 weeks [170].
Surprisingly, little change in glucose synthesis was identified in the rats treated by AO alone, and it was accompanied by elevated glucagon levels and an 80% reduction in glucagon clearance. However, this phenomenon was prevented when the rats received somatostatin or glucagon-neutralizing antibody, together with the AO. The combined treatments led to significantly decreased glucose production [170]. In addition, AO-induced CRTC2 reduction also resulted in decreased expression of enzymes participating in the conversion of amino acids to gluconeogenic intermediates (pyruvate and oxaloacetate), such as pyruvate carboxylase, glutamic-oxaloacetic transaminase 1, and serine dehydratase [170]. This research suggests that CRTC2 reduction could lead to improved hyperglycemia if combined with glucagon inhibition.
Dullea et al. presented a chemically modified AO development process using CRTC2 as a model [171]. They designed and synthesized around 400 CRTC2-specific 3–8–3 LNA-PS gapmers and performed a three-step screening process: first, transfection of AOs into both human (Hep3B) and mouse (Hepa1–6) cell lines was conducted to select candidates inducing efficient CRTC2 mRNA knockdown; second, 41 AO “finalists” were then evaluated in vivo in mice to determine both hepatic CRTC2 mRNA reduction and safety profile; and third, the “champion” and “runner-up” AOs were examined in DIO mice and human primary hepatocytes for subchronic efficacy studies [171]. This research confirmed the beneficial effect of CRTC2 knockdown toward ameliorating glucose control and provides an example of development of target-specific gapmer AO through a step-by-step selection process.
Forkhead box O1
Fasting hyperglycemia is mainly due to raised gluconeogenesis. FOXO1 is a primary transcription factor involved in gluconeogenesis that promotes the expression of PEPCK and G6P [172,173]. Reduction of FOXO1 in nucleus decreases the production of PEPCK and G6P, and thereby attenuates gluconeogenesis [174]. Samuel et al. proved that AO-induced FOXO1 reduction improves glucose control [175].
In that study, an FOXO1-specific 5–10–5 2′-OMOE-PS gapmer (ISIS 188764) was utilized to transfect mouse primary hepatocytes with an AO concentration of 100 nM, and DIO mice was administered with this AO at a dose of 50 mg/kg/week for 4 weeks [175]. Markedly decreased PEPCK (−48%) and G6P (−64%) in hepatocytes were observed, compared to the untreated group. In DIO mice, AO treatment led to significant reduction of fed plasma glucose level (−34%) and insulin concentration (−87%), indicating an improved insulin action. In addition, ameliorated insulin resistance was also identified in both liver and fat that correlated with decreased hepatic triglyceride and diacylglycerol content, and suppression of lipolysis in adipocytes [175]. This research suggests that AO-induced FOXO1 reduction could be a potential therapy for insulin resistance and T2D.
Density-enhanced phosphatase-1
The activity of IR kinase determined by phosphorylation status is regulated by PTPs, including PTP1B [176]. PTP1B also directly deactivates IR other than IR kinase by dephosphorylating its tyrosine residues [177,178]. Comparable to PTP1B, DEP1, as a phosphatase, may interfere with insulin signaling through also dephosphorylating IR. To examine the role of DEP1 in the insulin signal transduction pathway, Krüger et al. treated mice with HFD-induced insulin resistance with a DEP1-specific 5–10–5 2′-OMOE-PS gapmer AO (ISIS 285564) by IPI at a dosage of 25 mg/kg (twice a week) for 6 weeks [179].
AO treatment resulted in significant body weight loss, involving reduced epididymal and perirenal fat weight. Importantly, AO-induced DEP1 suppression improved insulin sensitivity that was reflected by significant decreases in baseline glucose level and fasting insulin concentration, accompanied by hepatic hyperphosphorylation of insulin signaling intermediates, including IR and Akt [179]. This study revealed the role of DEP1 as an endogenous antagonist of insulin signaling elements, especially IR. Therefore, reduction of DEP1 represents a novel therapeutic strategy for insulin resistance and T2D.
Retinol binding protein 4
RBP4 is an adipocytokine associated with hepatic steatosis and insulin resistance [180–184]. Increased serum RBP4 level causes insulin resistance by activation of proinflammatory pathways in fat tissues [185–187]. In fact, the level of circulating RBP4 is positively correlated with the magnitude of insulin resistance in obese T2D patients [188–190].
In an attempt to investigate the role of overexpressed RBP4 in the development of T2D, Tan et al. treated HFD-fed mice with an RBP4 targeting 2′-OMe-modified 18mer AO (25 mg/kg, IPI, once every 2 days) for 4 weeks [191]. Mice fed by HFD showed markedly increased weight of visceral fat, elevated levels of circulating triglyceride, FFA, glucose, and insulin, reduced GLUT4 expression, and increased PEPCK expression, suggesting an induced insulin-resistant state, whereas anti-RBP4 AO treatment avoided these changes. In addition, histological observation revealed that HFD-induced fat infiltrations and hepatic steatosis were prevented by anti-RBP4 AO treatment [191]. This study indicated the association of RBP4 with NAFLD and T2D, and reduced expression of RBP4 by AO treatment could be efficacious for T2D.
Transthyretin
TTR is a transport protein of RBP4 and thyroxine [192]. TTR binding to RBP4 decreases the glomerular filtration rate of RBP4, thereby retaining it in the blood [193]. Therefore, reduction of TTR lowers RBP4 levels by increasing renal clearance, and subsequently ameliorates insulin resistance. To determine whether reduction of TTR by AOs improves glucose homeostasis and insulin signaling, Zemany et al. administered a TTR-specific 5–10–5 2′-OMOE-PS gapmer AO to ob/ob mice (ISIS 401723, 12.5 mg/kg IPI) and DIO mice (ISIS 401724, 25 mg/kg SCI) twice a week, respectively [194].
Two weeks of AO treatment led to dramatic reductions in circulating RBP4 levels in ob/ob mice and DIO mice by ∼80% and ∼95%, respectively, which was associated with 45%–60% increase in muscle glucose uptake, decreased glucose production in liver, and reduced fat inflammation. These changes correlated with a marked increase of insulin signal transduction in muscle by twofold, and 30%–60% reduction in insulin levels that were sustained for 9 weeks, suggesting increased insulin sensitivity [194]. The study revealed that AO-induced TTR reduction could be a potential therapeutic strategy for the treatment of insulin resistance and T2D through lowering RBP4.
Tribbles homolog 3
Overexpressed TRIB3 is associated with insulin resistance and T2D as TRIB3 negatively regulates Akt in the insulin signaling pathway [195,196]. On the other hand, TRIB3 inhibits the activity of peroxisome proliferator-activated receptor γ (PPARγ), and thus suppresses adipogenesis [197]. Activation of PPARγ is associated with improved insulin signal transduction that leads to increased insulin sensitivity [198], as the induction of adipogenesis (resulting from PPARγ activation) associated with the capability for FFA trapping has been shown to be a vital contributor to the systemic maintenance of insulin sensitivity.
Weismann et al. explored the role of TRIB3 reduction in improving insulin resistance by administering HFD-fed rats with a TRIB3-specific 5–10–5 2′-OMOE-PS gapmer (ISIS 391274) at an IPI dose of 75 mg/kg weekly for 4 weeks [199]. AO treatment led to ∼50% increase in the rate of whole-body glucose uptake under euglycemic-hyperinsulinemic condition that was mainly attributed to the raised muscle glucose uptake. Moreover, ISIS 391274 treatment relieved the inhibitory effect of TRIB3 on PPARγ and its downstream enzymes/transcription factors, such as acyl-CoA oxidase and CCAAT/enhancer binding protein α, leading to increased level of these proteins, raised adiponectin levels, and expanded white fat tissue mass by ∼70% [199].
It is noteworthy that when PPARγ was inhibited by an antagonist, the aforementioned changes resulting from AO treatment were prevented, suggesting that the improved insulin resistance resulting from TRIB3 reduction is PPARγ dependent [199]. Based on this study, inhibition of TRIB3 might be beneficial for the therapy of insulin resistance and T2D.
IκB-kinase β
Chronic inflammation associated with obesity is a major contributor to T2D [200,201]. Overnutrition results in IκKβ activation [202–204], which subsequently activates NFκB, a transcription factor that regulates inflammatory pathways contributing to insulin resistance [205,206]. Therefore, IκKβ could be a potential target for the therapy of obesity-associated T2D. To this end, Helsley et al. explored AO-based IκKβ inhibitor as a pharmacological approach to insulin resistance and obesity by treating DIO mice with an IκKβ targeting 5–10–5 2′-OMOE-PS gapmer at a biweekly IPI dosage of 25 mg/kg for 8 weeks [207].
AO treatment led to reduced levels of fasting plasma glucose and insulin, and increased Akt phosphorylation and glucose uptake in fat, suggesting improved insulin sensitivity [207]. Furthermore, the IκKβ targeting AO significantly decreased adiposity and weight gain, with a 45% reduction in fat mass that correlated with decreased hepatic triglyceride content, diminished adipocyte differentiation from adipose stromal vascular cells, and the fact that lipid accumulation and hepatic steatosis were avoided in DIO mice [207]. These findings establish IκKβ as a vital regulator of adipogenesis, and reduction of IκKβ by AO represents a potential therapeutic approach to combat insulin resistance and obesity.
Toll-like receptor 2
TLRs are associated with upregulation of inflammatory cytokines through activation of NFκB [208,209]. Inflammation mediated by cytokines such as TNFα and IL6 eventually leads to insulin resistance [36–38]. Senn showed that activation of TLR2 inhibited insulin signaling pathway in differentiated C2C12 myotubes [210].
To investigate the effect of TLR2 reduction by AO treatment in HFD-induced insulin resistance, Caricilli et al. used a TLR2 targeting 18mer AO to treat DIO mice at an IPI dose of 25 mg/kg (mice were killed 4 days after single AO injection) [211]. It is known that activation of IκKβ and JNK1 induces insulin resistance through IRS1 inhibition [212], and TLR2 AO treatment led to decreased phosphorylation of IκKβ and JNK1 in both muscle and fat, suggesting an improvement to insulin resistance [211]. Furthermore, increased phosphorylation of IRS1 and Akt was also identified in muscle and fat, indicating increased IRS1 and Akt activation and ameliorated insulin signal transduction [211]. This study revealed that AO-induced reduction of TLR2 improved insulin resistance in peripheral tissues [211], thereby highlighting the potential of TLR2 as a target for T2D therapy.
Clinical Trials
To date, none of the AOs developed as inhibitors of genes involved in the T2D pathogenesis has been approved for clinical application. This might be due to the fact that, unlike monogenic diseases such as DMD, a number of genes are involved in T2D pathogenesis and might thereby collectively contribute to the onset and progression of T2D. In contrast, a number of small-molecule hypoglycemic agents have been approved and commercialized as T2D therapeutics (Table 2). Comfortingly, some investigational AO candidates have entered clinical trials and they are detailed in Table 2.
Approved Small-Molecule Drugs and Investigational Antisense Oligonucleotides in Clinical Trials Targeting Insulin Resistance and/or Hyperglycemia in Type 2 Diabetes
SGLT2, sodium glucose cotransporter-2; DPP4, dipeptidyl peptidase-4; PTP1B, protein tyrosine phosphatase-1B; GCCR, glucocorticoid receptor; DGAT2, diacylglycerol acyltransferase-2; APOC3, apolipoprotein C-III; ANGPTL3, angiopoietin-like protein 3.
Conclusion
Traditional T2D therapy has largely relied on small molecule-based oral agents aiming at reducing blood glucose level by targeting essential glycometabolic organs and tissues. However, these agents do not halt the progression of T2D due to limited efficacy, and they induce adverse reactions that affect patient compliance as well.
Alternatively, efforts made on identifying genes involved in the pathogenesis of insulin resistance and hyperglycemia underlying T2D lead to the emergence of a pragmatic treatment option: direct inhibition of the disease-causing genes on either protein or RNA level. This is achieved by developing small molecule- or antibody-based protein inhibitors, which bind to and block the activity of target proteins, or nucleic acid-based agents that interfere with the transcriptional or translational process of target genes, thereby reducing the production of those proteins.
Although some small molecule-based protein inhibitors have been approved for T2D therapy (Table 2), the development of protein inhibitors is facing obstacles in terms of druggability, selectivity, and toxicity due to the fact that many proteins lack binding pockets for small molecules, the diversity of protein isoforms limiting drug specificity, and potential interaction of drug molecules with nontarget biomacromolecules.
In contrast, nucleic acid-based intervention possesses highly competitive advantages: therapeutic oligonucleotides reduce the production of target proteins by inhibiting gene expression (transcription/translation) and thus obviate directly coping with the undruggable proteins; oligonucleotides selectively and specifically bind to mRNA transcript of interest, thereby avoiding inhibition of nontarget protein isoforms and off-target effects; the diversity of antisense mechanisms (RNase H induction, splice modulation, and translational arrest shown in Fig. 2), chemical modifications (PS, 2′-OMe, 2′-OMOE, 2′-F, LNA, ENA, PMO, PNA, etc. shown in Fig. 3), and design strategies (uniform modification, gapmers, mixmers shown in Fig. 4) provides sufficient potential options and tools in the optimization of AO candidates for improving their efficacy, stability, and safety; AO-induced gene inhibition is useful in the identification, functionalization, and validation of target genes implicated in T2D pathogenesis, and the AOs themselves are directly employed as candidates in the simultaneous or subsequent drug development process; therefore, AO is a versatile platform that plays central roles in both target discovery and drug development for T2D.
In fact, given that the recently increasing approvals of AO-based therapeutics in tackling various human diseases (Table 1), and some AO candidates developed for tackling T2D have entered clinical investigations of different stages (Table 2), we firmly believe that approvals of antisense-based antidiabetic agents for clinical use will become realistic in the near future.
Footnotes
Acknowledgments
R.N.V. acknowledges the financial support from McCusker Charitable Foundation and Perron Institute for Neurological and Translational Science. S.C. thanks the funding from Perron Institute Top-Up Scholarship and International Tuition Fee Scholarship scheme from Murdoch University.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by funding from Perron Institute for Neurological and Translational Science and McCusker Charitable Foundation.