
Abstract
The Oligonucleotide Working Group of the European Federation of Pharmaceutical Industries and Associations (EFPIA) conducted a survey of companies to understand the trends in nonclinical practices and regulatory expectations for oligonucleotide drug safety assessment. Twenty-two companies of different types, with varying oligonucleotide experience levels in the field, participated. The survey identified key regulatory challenges and areas of perceived health authority (HA) concern regarding nonclinical safety strategies for oligonucleotides, such as the choice of toxicology species, approaches to dose setting in toxicity studies, dose scaling from animals to humans, the implementation (and regulatory acceptability) of lean packages, and methods for dealing with impurities and human-specific off-targets. The perceived oligonucleotide experience of HAs and the relevance of guidance to oligonucleotide development were also assessed. The results showed a general lack of consensus on nonclinical safety assessment approaches being used for this growing class of medicines and highlight the need for continuing collaboration between sponsors and HAs to better define best practices.
Introduction
With an intermediate molecular weight, oligonucleotide-based drugs are distinct from both small molecules and biologics [1]. On one hand, nucleic acid therapeutics are similar to biologics in that they mimic naturally occurring mechanisms found in every living cell and that they can be highly specific for their target. On the other hand, most therapeutic oligonucleotides contain chemically modified backbones and/or ribose sugars to enhance their in vivo stability and potency. Based on their chemical synthesis, they are currently regulated as small-molecule new chemical entities.
An Oligonucleotide Nonclinical Safety Working Group (WG) was established in 2017 under the sponsorship of the European Federation of Pharmaceutical Industries and Associations (EFPIA). The development of oligonucleotides as drugs started back in the 1990s [2]. With hindsight from three decades of experience and a focus on nonclinical safety assessment, the members of this WG set out to survey the industry on the trends in nonclinical practices and regulatory expectations, as well as potential issues experienced in the development phase for oligonucleotide safety assessment. The group designed an online questionnaire and sent it out to companies developing therapeutic oligonucleotides in the United States, Europe, and Japan. The WG was interested in querying sponsors on various issues, including typical regulatory feedback and the outcome thereof, suitability of existing international guidelines to the needs of oligonucleotide drug development, and forward-looking views on modified safety strategies for oligonucleotides.
Highlights from this survey were presented and discussed at the Drug Information Association (DIA)/Food and Drug Administration (FDA) Oligonucleotide-Based Therapeutics Conference in Bethesda, Washington DC, in October 2019. Based on the survey results and the discussion at the conference, there is a general lack of clarity on many issues pertaining to oligonucleotide development among both sponsors and health authorities (HAs). Increased clarity is desired by most stakeholders but whether this should be in the format of research-based publications, best practice articles, or formal guidelines continues to be a matter of debate.
Materials and Methods
Nine EFPIA member companies assigned a representative (see author list) with F. Hoffmann-La Roche Ltd. leading the WG. The survey used a commercial online survey application (SurveyGizmo®), which was administered by the EFPIA secretariat to secure blinding of the WG to respondents' identity (WG members were also invited to respond). The survey was structured as follows:
Questions aimed to categorize the company type, its level of oligonucleotide development experience, and its oligonucleotide portfolio maturity. Portfolio-level questions (23 questions and 220 items in total, ie, subquestions or options) covering the areas of: Nonclinical safety (main focus) Chemistry Manufacturing and Controls (CMC) Clinical. Optional: case studies describing regulatory interactions for oligonucleotide projects (up to 10 per respondent).
As inclusion criteria, respondents had to be employed by an EFPIA member or nonmember company, with at least one therapeutic oligonucleotide asset in their current pipeline from pre-First in Human (FiH) Good Laboratory Practice (GLP) phase through marketing stage. For the purposes of this survey, the oligonucleotide definition excluded messenger RNA (mRNA) mimics, gene editing, or other advanced modalities not regulated as small molecules (Table 1). They also had to have received regulatory feedback on the nonclinical package for at least one program. To prevent data duplication from potential cross-company collaborations, respondents working on the same project were asked to assign a single respondent for the project concerned.
Oligonucleotide Modalities Meeting the Inclusion Criteria
CDER, Center for Drug Evaluation and Research; lncRNA, long noncoding RNA; mRNA, messenger RNA; siRNA, small interfering RNA.
A pilot version of the survey was used among the WG members to check for question relevance, logical operators, overall acceptability of volume/time to complete, and typographical errors.
Thirty companies (including those of the WG members) operating in the United States, Europe, or Japan were preidentified as in-scope and were invited to participate in the definitive survey between May 2, 2018, and July 31, 2018. Twenty-two anonymized, filled questionnaires were received. Further 33 blinded case studies were submitted. The content of the case studies typically overlapped with responses provided in the general questionnaire and was used by the authors to provide additional context and strengthen interpretation of responses to the closed questions.
The results were analyzed, discussed, and summarized by the WG members. Due to the small sample size, no statistical analysis was performed.
Results
Respondent statistics
Three company types could be selected for self-categorization (“biotech,” “small-mid pharma” and “large pharma”). Although dominated by “biotech” (∼41%), the distribution was overall balanced between those and “small-mid” (∼23%) and “large pharma” (∼32%) companies (Fig. 1). A single company used the free-text option and referred to themselves as a “chemicals company.” Figure 1 also shows that only 3/22 companies had an oligonucleotide development portfolio of 5 or more assets at the time of the survey.
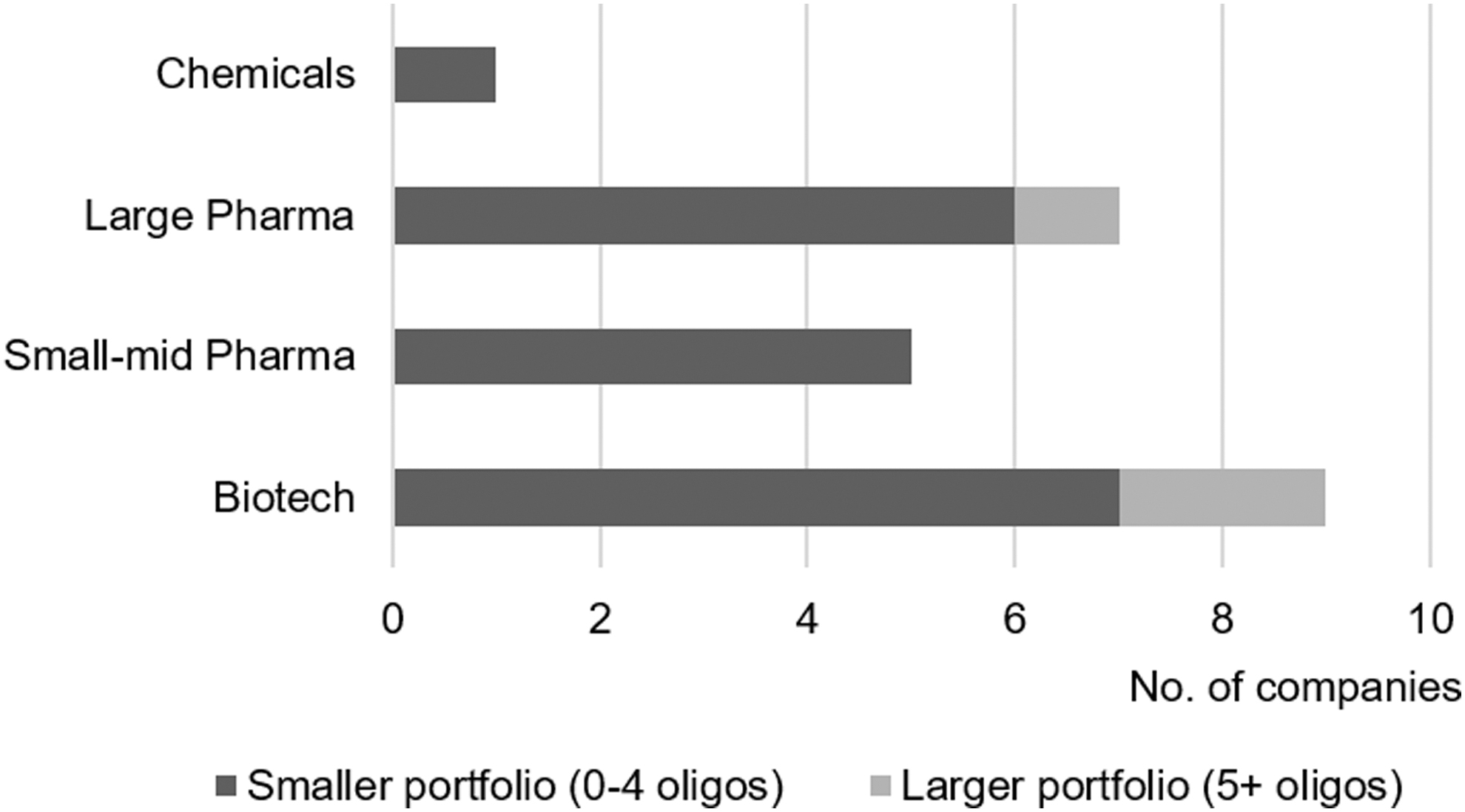
Company size and oligonucleotide portfolio maturity.
The definition of oligonucleotides encompassed diverse subclasses, as shown in Table 2, with double-stranded small interfering RNA (siRNA; 8 of 22 respondents) and single-stranded RNase H-dependent (6/22) designs being the most commonly reported.
Subclasses Represented in Respondent Portfolios
Some companies had more than one subclass in their portfolio, hence the number >22.
Based on responses to the question “Please tick the stages reached by your oligonucleotide candidate(s),” the cumulative pipeline size of all respondents amounted to at least 47 assets, with balanced distribution through the pre-FiH to postmarketing stages (Fig. 2).
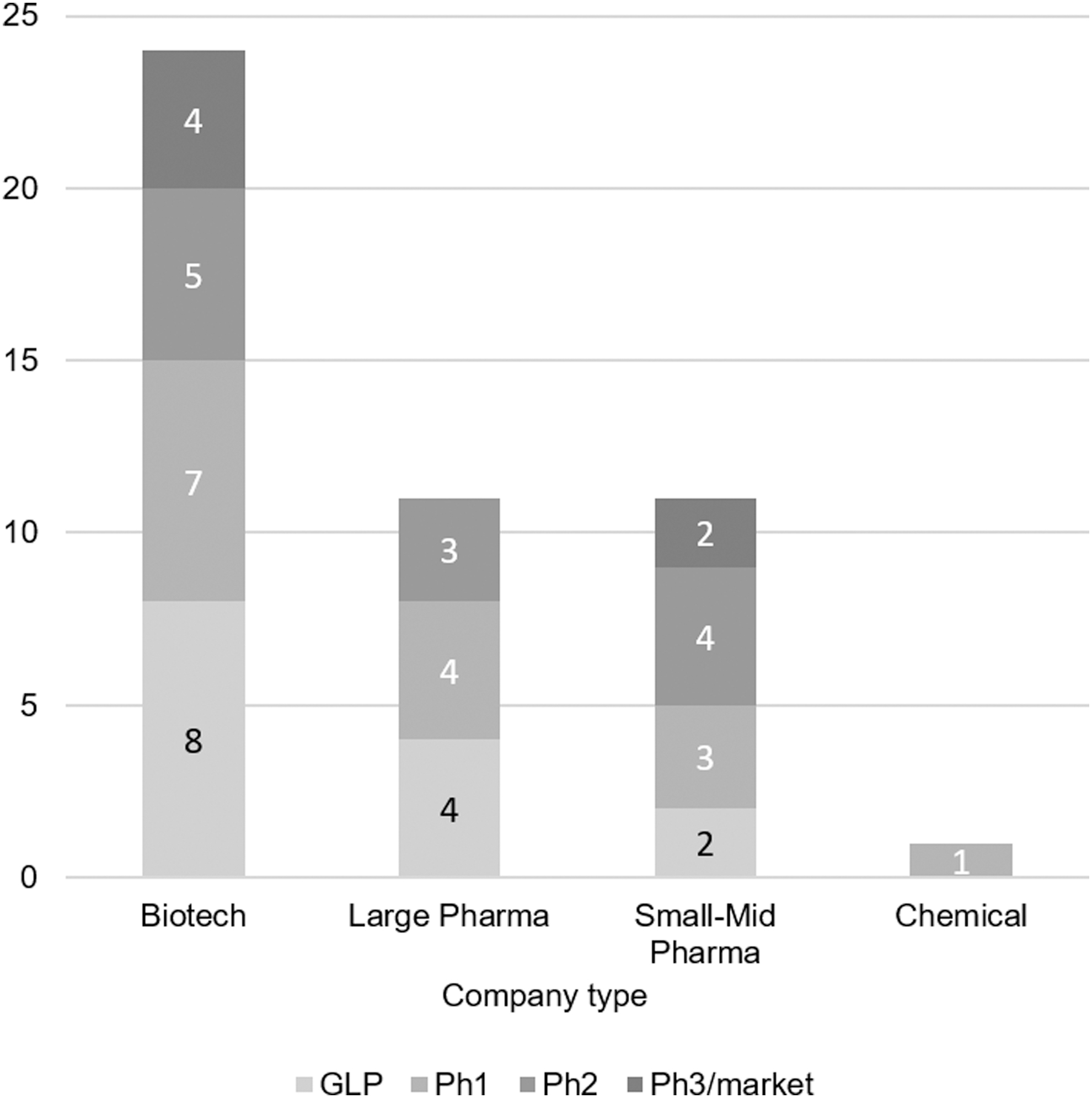
Development pipelines: number of assets versus phase.
The prevailing routes of administration used by the respondents were parenteral (ie, subcutaneous and intravenous), followed by local (defined in this survey as intravitreal, intrathecal, or intralesion), topical (eg, eye drops, oral/rectal, or dermal), and finally, respiratory (which, due to its specific nature, was kept separate from the other topical routes) (Fig. 3).

Number of respondents using the following routes of administration.
Trends in HA feedback
The questionnaire then sought to understand absolute numbers of HA interactions, per country, as well as the top-five regulatory challenges encountered by the respondents for their oligonucleotide projects.
Table 3 shows that according to the sponsors that responded to the survey, the US FDA, followed by major agencies from Europe and Japan, is viewed as having the most exposure to oligonucleotide-related submissions, in absolute terms (ie, not normalized to agency size). With respect to the FDA, it is generally acknowledged that oligonucleotide experience varies across divisions. A subquestion was therefore asked to understand if sponsors considered being treated inconsistently across divisions, to which only 29% replied they did.
Exposure of Regulatory Agencies
EMA, European Medicines Agency; FDA, Food and Drug Administration; HA, Health Authority.
In decreasing order of frequency, and based on free-text responses, the main regulatory challenges faced by oligonucleotide sponsors were (Table 4) as follows: (1) variable knowledge about oligonucleotide drugs/lack of oligonucleotide-specific regulatory guidelines; (2) CMC issues, in particular impurity control practices; (3) management of low safety margins, especially when based on rodent-specific class effects; (4) accepted approaches for preclinical to clinical dose scaling; and (5) increasing HA queries about off-target effect (OTE) evaluations.
Top Regulatory Challenges (Bucketed Based on Free-Text Responses)
CMC, Chemistry Manufacturing and Controls; OTE, off-target effect.
Respondents were then asked to rank their perception of topics of concern to HAs regarding oligonucleotide drug candidates. The first options here were predefined, and a free-text field was offered for any additional suggestions. The prevailing CMC regulatory concern (as perceived by sponsors) was the issue of product-related impurities and their qualification (Table 5). For nonclinical safety questions, the category of most common concern was “nonclinical development strategies supporting clinical development or marketing” (Table 6). Free-text responses here included examples of the required nonclinical safety studies to support various stages of clinical development, pharmacokinetic evaluation practices, toxicology species selection, study duration, study package content, and timing of studies relative to the clinical program.
Oligonucleotide-Specific Perceived Topics of Concern to Health Authorities (Chemistry Manufacturing and Controls)
Oligonucleotide-Specific Perceived Topics of Concern to Health Authorities (Nonclinical)
For questions relating to clinical studies (Table 7), “clinical dose setting” was the prevailing perceived concern of HAs. Among the free-text responses, methods used for dose scaling from animals to humans, human starting dose selection, and human dose capping were key themes.
Oligonucleotide-Specific Perceived Topics of Concern to Health Authorities (Clinical)
ADA, antidrug antibody.
The next question was intended to assess the impact that HA feedback had had on respondent's oligonucleotide projects (Table 8). The most common nonclinical regulatory package follows the principles of the two-species ICH M3(R2) guideline [3]. For 29% of sponsors, HAs had (on at least one occasion) accepted a leaner M3(R2) approach adapted for oligonucleotides upon first submission. Seventy-six percent of the respondents had at least once received requests from an HA for a study or a readout, which they had not included or planned to include (see Table 8, eg). However, 47% of the respondents had experienced situations where the HA, after a dialogue, eventually accepted the sponsor's rationale for a “leaner,” oligonucleotide-tailored package that differed from the default two-species ICH M3(R2)-type approach. Responses to specific questions included 53% of sponsors having received requests for antidrug antibody (ADA) assessment. Interestingly, the development stages at which these requests were made ranged from GLP toxicity to Phase 2, as can be seen in Table 8. Moreover, for 18% of respondents, HAs had requested (at least once) capping a clinical dose in a noncancer indication based on the existing nonclinical safety information (a free-text comment cited the long half-life of the molecule as the primary concern). Finally, only 12% of the respondents encountered a situation where an HA request had a significant impact on, for example, time lines or cost of a project.
Percentage of Respondents Having at Least Once Received Health Authority Feedback Where…
CTA, clinical trial application; ePPND, enhanced pre- and postnatal development; GLP, Good Laboratory Practice; Ph1/2, Phase 1/2; t1/2, half-life.
The last question in this section was on perceived oligonucleotide experience of agencies throughout the world, based on sponsor rating from 0 to 100. Figure 4 is a three-dimensional plot of experience rating versus number of HA interactions versus number of responses from which the average rates were calculated. Focusing on the ratings above 50%, sponsors have had the most experience with submissions and follow-up interactions with US FDA and rated this HA among the highest (note: the survey did not offer to break down ratings by division). Although less exposed (as per absolute number of sponsor interactions and responses), both major and smaller European agencies cluster together with Pharmaceuticals and Medical Devices Agency (PMDA, Japan) as highly rated by oligonucleotide sponsors.

Perceived oligonucleotide experience of health authorities. Color images are available online.
Toxicology testing strategies for oligonucleotides
Acknowledging that indications and oligonucleotide subclasses differ in their requirements, the lack of consensus on the best nonclinical strategy for the development of oligonucleotide-based drugs is not surprising. To get a better understanding of the current thinking in the industry, respondents were asked questions on the design of toxicology programs for oligonucleotides. The majority of respondents (76%) have always conducted toxicology testing in two species, which is consistent with the ICH M3(R2) requirements for pharmaceuticals [3]. This was the case for both single-stranded and double-stranded oligonucleotides (Table 9). In instances where single-species nonclinical safety programs were conducted, they were primarily done using nonrodent species based on the following: (1) lack of cross-reactivity to the pharmacologic target in rodents, (2) lack of feasibility of the clinical dose route in rodents (eg, intrathecal dosing), and (3) low risk of systemic class-related or off-target toxicity based on route of administration. Based on the case studies provided by respondents, single-species toxicology packages have generally been accepted by HAs when accompanied by sufficient justification. The survey data also showed that smaller companies are more willing to use single-species toxicology packages, while none of the respondents identified as large pharma has ever used this approach (Table 10).
Number of Respondents Having Used Single-Species Packages Based on Oligonucleotide Design
Number of Respondents Having Used Single-Species Packages Based on Company Type
A second question was related to species selection for toxicity studies. A little over half of the respondents (55%) used rat as the default species versus 27% that defaulted to mouse (Table 11). The remaining respondents selected the rodent species on a case-by-case basis. When broken down by subclass, though, use of mouse prevailed for single-stranded oligonucleotide candidates, while rat was the main rodent species used in double-stranded oligonucleotide projects. For the nonrodent species (Table 12), 52% had only ever used the nonhuman primate (NHP) regardless of oligonucleotide class. However, there were respondents who have used dog, rabbit, or swine although the responses did not specify whether this was in addition to or instead of NHP. The choice of the nonrodent toxicology species was driven by a number of factors, including the route of administration, biologic activity of the oligonucleotide in a given species, and, in two cases, to assess the toxicology profile in the nonrodent species (rabbit) that was used for the nonclinical efficacy/pharmacology studies. The respondents who reported they only had used NHPs before were asked whether they would consider alternative nonrodent species in the future. Among those who responded, 62% (5 of 8) were open to it. Criteria mentioned as free-text included the 3R principles (Replacement, Reduction, and Refinement), cost, NHP sourcing issues, and, once more, cross-species homology.
Rodent Species Selection
Prior Use of Nonrodent Species According to Subclass
ds, double-stranded; NHP, nonhuman primate; ss, single-stranded.
Toxicity study dose selection
The survey results revealed variability in how sponsors select the high doses for the toxicity studies in noncancer indications (Fig. 5). Twenty-four percent of the respondents target the highest possible dose, whether it be maximum tolerated dose (MTD) or maximum feasible dose (MFD). The majority of respondents (66%) relate to what they deem to be “pharmacologically relevant exposures” when setting the top toxicology doses. Upon further probing, the definition of “relevant” differs among respondents: as shown in Fig. 5, among those favoring an exposure multiple approach, a little under half (45%) indicated they target a 10-fold multiple of the predicted clinical exposure, with the remainder targeting 25-fold (33%), 50-fold (11%), or taking a case-by-case approach (11%). The approach to set the top dose level did not markedly differ between single-stranded and double-stranded designs, although a 50-fold margin was only mentioned for the latter (data not shown). In general, these margin-based approaches appear to have been accepted by global HAs. However, there was one example provided where an HA did not agree to setting the high dose based on a 10-fold margin and instead requested a 50-fold margin [consistent with ICH M3(R2)] [3].
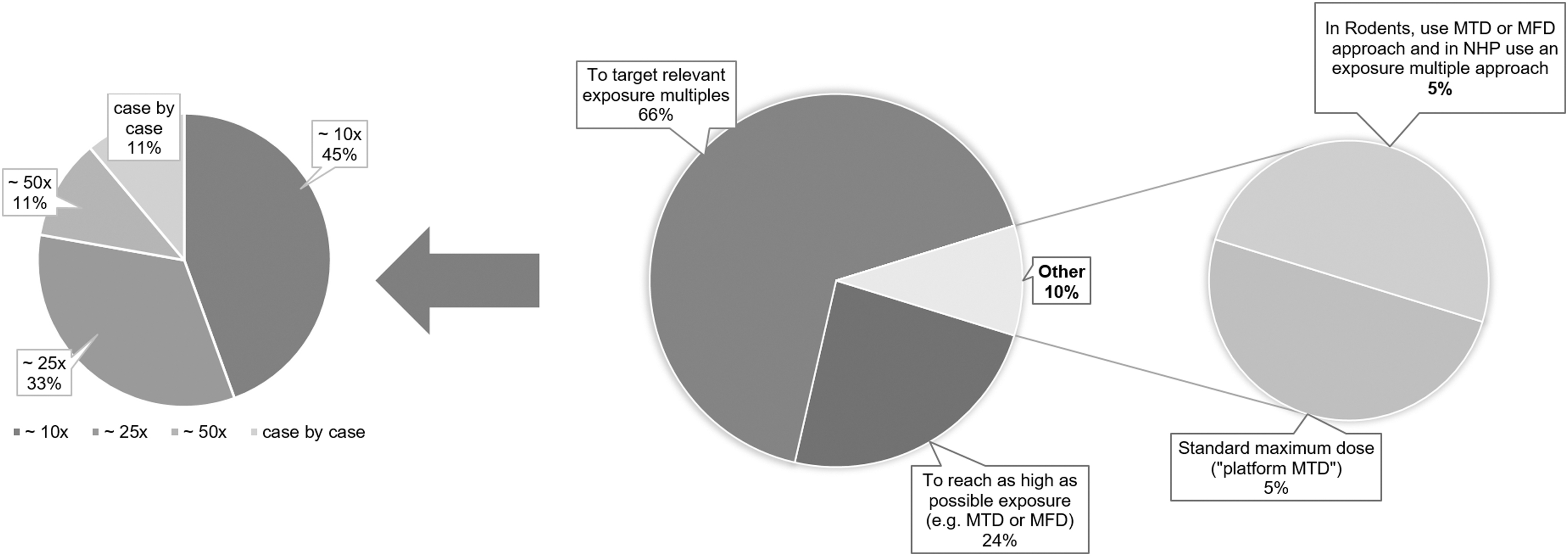
High dose selection in GLP toxicity studies (noncancer indications) and exposure multiple targets. GLP, Good Laboratory Practice.
Scaling dose levels from animal to human
A topic related to toxicology dose selection is the approach used to translate animal dose to human dose when calculating safety margins. As shown in Fig. 6, there is a lot of variability in how respondents do this dose scaling and this differs based on the animal species and oligonucleotide class. For instance, between NHPs and humans, for systemically administered oligonucleotides, 44% of the respondents scale doses 1:1 on an mg/kg basis while 33% scale doses based on body surface area (BSA), with no clear difference between single-stranded and double-stranded oligonucleotides. In contrast to NHP studies, BSA-corrected scaling is the most common approach in both rats and mice for double-stranded oligonucleotides among the respondents. The most variability was seen in approaches to scale single-stranded oligonucleotide doses from rodents to humans, with scaling factors ranging from 1 × to 10 × for rats and >12 × for mice.
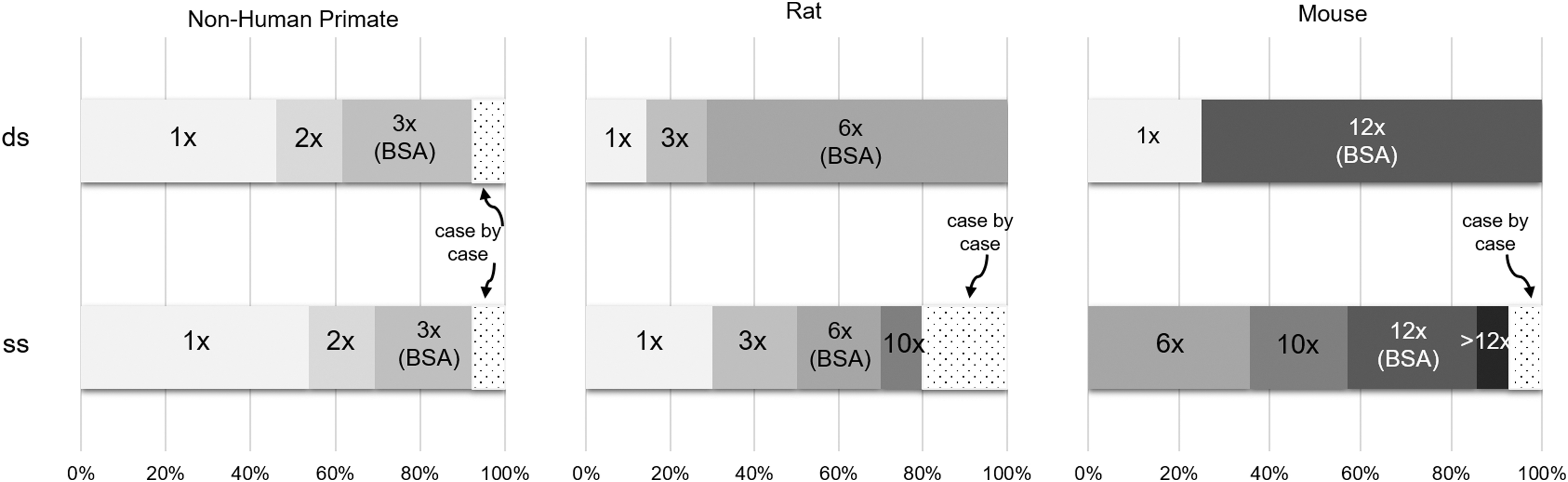
Targeted exposure multiples as a function of oligonucleotide design and species (systemic routes).
Off-target effects
A free-text response was requested to the question “How do you deal with human-specific hybridization off-targets?” In general, most respondents outlined approaches consistent with that described in the Oligonucleotide Safety Working Group (OSWG) 2012 article [4], which includes in silico prediction of off-target binding, in vitro verification to define relative potency, and risk assessment based on understanding of the off-target biology and function. Three of 22 respondents reported current or planned future use of global transcriptome analyses (eg, RNASeq) to evaluate OTEs.
Genetic toxicity assessments
Part of the questionnaire dealt with the option of using innovative or lean study packages, tailored to oligonucleotide drug candidates. When asked which studies or readouts companies had successfully removed from their packages, four respondents (three “biotech” and one “small-mid pharma”) reported they had run preclinical packages with no or reduced testing of genetic toxicity.* To the authors' knowledge, no therapeutic oligonucleotide of established chemistry and mechanism of action has been reported to be positive in a standard genetic toxicity assessment [5–7], but the ICH S2(R1) guideline [8] does not explicitly exclude running standard batteries of genetic toxicity for oligonucleotide therapeutics. Fifteen out of 22 respondents implied in a free-text response on waiving genetic toxicity assays that these should be waived entirely (including for noncancer indications), at least for established chemistries. When asked about the relevance of ICH S2(R1), 7/22 respondents allotted 0% relevance to the guidance, while overall mean relevance was only 34%.
Impurity management
Impurity management for oligonucleotides is an area of active discussion. Survey respondents considered the set of ICH Q3 guidelines [9,10] (which cover a broad range of impurity topics) to be 61% relevant on average. Following the community reception of a recent article describing alternative thresholds for impurity qualification and quantification for oligonucleotides [11], the survey asked “In the future, which qualification threshold will you apply, as default?” As shown in Fig. 7, 14/22 respondents indicated they would apply the 1.5% impurity threshold proposed in the article by Capaldi et al. [11] as a default compared with 1/22 who indicated they would use the 0.15% impurity threshold as outlined in ICH Q3. Based on the narratives in the case studies included in this survey, interactions with HAs regarding impurities generally resulted in sponsors qualifying impurities by demonstrating coverage in toxicity studies rather than making a case for applying the different impurity standards for oligonucleotides proposed by Capaldi et al. [11].
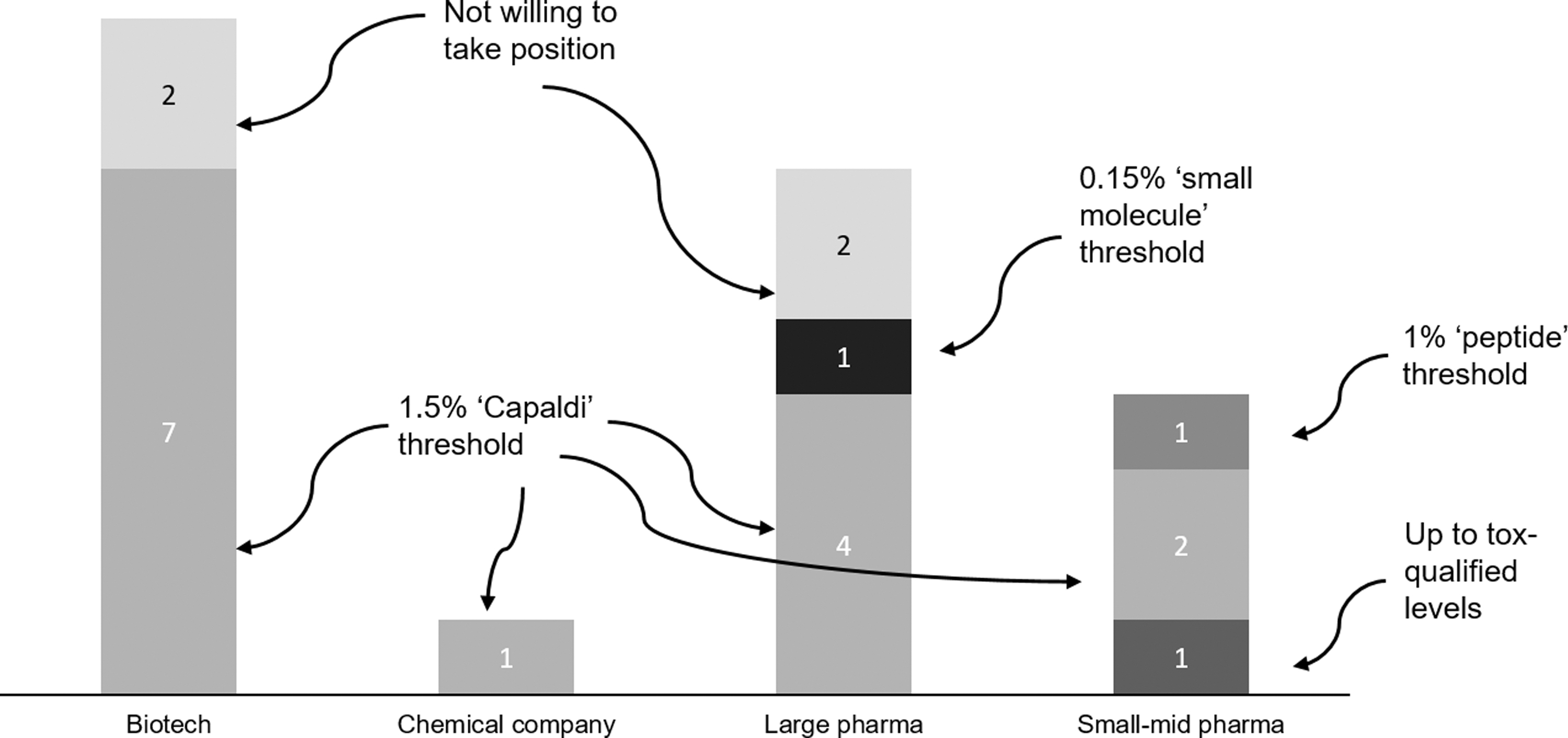
In the future, which qualification threshold will you apply, as default?
Relevance of existing guidelines
In the absence of dedicated oligonucleotide guidance, HAs have so far defaulted primarily to small-molecule guidelines such as ICH M3(R2). Participants in this survey were asked, “To what extent do you deem the existing guidelines relevant to oligonucleotide development (scale 0–100)?” As shown in Fig. 8, the responses ranged from ICH M3(R2) at the high end (72% mean relevance) to the Metabolites in Safety Testing (MIST) at the low end (28% mean relevance). With the exception of ICH M3(R2), all guidelines had at least one response of 0% and at least one of 100%. ICH S6(R1) [12] is the nonclinical guidance for biologic products but does state that the principles outlined may be applicable to oligonucleotide drugs. Respondents placed this on the lower end of relevant guidelines with an overall mean relevance of 44%. When respondents were stratified by reported experience, those who described themselves as “very experienced” considered S6(R1) less relevant (20%) than those who described themselves as “novel to oligonucleotide drugs” (60%) (data not shown).

Ranked reported relevance of existing guidelines. Color images are available online.
Need for new guidelines
Respondents were asked about the need for new formal guidelines that would specifically address issues common to oligonucleotide development. Respondents could choose multiple responses from the following: no new guideline or tailoring, support for tailoring existing guidelines for oligonucleotides, support for a new all-round oligonucleotide-specific impurity control guideline, and support for a new all-round oligonucleotide-specific nonclinical safety guideline. Fifteen of 22 respondents supported an oligonucleotide-specific impurity control guideline, while 11 and 12 of 22 supported tailoring or generating new guideline in other areas, respectively. Only one respondent thought no new guideline or tailoring was needed.
Survey participants were asked (as free-text response) about their rationale for their position on new guidelines and the main arguments for and against are summarized (Table 13).
Pros and Cons of Creating New Oligonucleotide-Specific Guidelines
3R, Replacement, Reduction, and Refinement.
When asked (as free-text responses) which kinds of guidelines should be prioritized in such an effort, the most frequently mentioned were addressing impurities, “general” nonclinical guidelines [note from authors: analogous to ICH M3(R2)] [3], and clarification of genetic toxicity testing requirements.
Discussion
The EFPIA-sponsored Oligonucleotide WG conducted a survey of companies actively developing therapeutic oligonucleotides, focused on understanding trends in both regulatory expectations and industry practices for their nonclinical safety assessment. Among the 22 respondents, the breadth of the industry was represented, with “biotech” companies unsurprisingly accounting for the greatest proportion. “Large pharma” and “small-mid pharma” altogether accounted for roughly half of the respondents, likely reflective of alliance partnerships with biotech companies that own oligonucleotide platforms. The full diversity of oligonucleotide subclasses were represented (eight categories in total), although siRNA and RNase H mechanisms were most frequently reported. The pipeline sampled included assets at varying phases of development and using various routes of administration.
The authors recognize that the survey sample represents but a proportion of the industry, with 22 companies responding. According to a database search, as of May 31, 2018, there were 31 sponsors developing Phase I through marketed nucleic acid drugs worldwide that met the subclass scope as per Table 1. The database † used does not enable retrieving GLP toxicity stage projects; however, it is reasonable to assume that a substantial part of the public portfolio has been captured. Setting up the survey, there was undoubtedly a bias toward companies that the EFPIA Oligonucleotide WG members were aware of; however, 30 companies from across Europe, the United States, and Japan were invited to participate and 22 responded. A noteworthy limitation of the survey is that for individual categories of company type and oligonucleotide subclass, samples sizes may have been too small to draw generalizable conclusions. However, with those caveats, it has certainly been possible to identify themes and trends, which may warrant further investigation.
One of the main goals of the survey was to identify trends in HA interactions, and several questions were tailored to probe this. The US FDA stood out in terms of frequency of interactions, with 19/22 respondents confirming they had made submissions to this HA for their oligonucleotide project(s). The European Medicines Agency (EMA), German and Japanese HAs each had approximately half the number of interactions noted for the FDA, with all others recording ≤6/22 companies each. These numbers are absolute and as such do not take into account the size of the agency in the respective countries: it could therefore be that individual staff members in smaller agencies have reviewed more oligonucleotide submissions than their counterparts in larger countries. When companies were asked to assign a “perceived level of experience” in oligonucleotide therapeutics to the HAs they had interacted with, those cited most frequently scored relatively high (ie, FDA, Germany, EMA, and Japan); however, that was by no means an exclusive indicator for a higher rating (eg, Sweden, the United Kingdom, Switzerland, Finland, France, Italy, and Canada all scored >50%). Other agencies did score less favorably in terms of “perceived experience”; however, a low number of respondents had had interactions with those, which may have reduced the reliability of the rating averages. Of note, for the FDA, only 29% of respondents considered that there was inconsistent treatment across divisions. It is certainly recognized that the FDA (among other agencies) has engaged positively with the industry for many years and has been proactive in educating reviewers, facilitated by the FDA's cross-division oligonucleotide subcommittee.
Some clear trends were identified when companies were asked to name their top five regulatory challenges. Variable knowledge of the modality, likely reflecting the earlier discussion around “perceived experience” of HAs, and a lack of oligonucleotide guidance ranked most highly. This theme had been anticipated by the survey authors, with a series of questions specifically addressing the relevance of existing guidance and the need for new guidelines (see later discussion). Others included CMC/impurity control practices, challenges related to low safety margins, and approaches for human dose scaling, as well as increasing focus of HAs on hybridization-dependent OTEs. Aspects of these themes were also reflected in responses when companies were asked to rank their “perceived topics of concern to HAs.”
In the section that focused on sponsor-HA dialogue, some notable trends were identified. Most companies (76%) had received initial requests from reviewing HAs for the conduct of additional endpoints/studies, although the survey does not enable pinpointing of development timing or the specific HAs represented here. Such examples included running studies in a second species or conducting additional genotoxicity assays, both of which would be considered standard for small-molecule drugs, but not for biopharmaceutical molecules. Nearly half (47%) of respondents indicated that they had experience (on at least one project) of HAs eventually accepting their rationale for a more streamlined nonclinical safety package, aligned to the scientific needs of the modality. This is encouraging evidence that agencies are open to a modified approach where there is a clear and compelling scientific rationale. The survey did not query whether HA willingness to accept modified packages in the oligonucleotide field differed from that in other modalities. Another notable trend was that over half (53%) of respondents indicated they had experience of HAs requesting ADA assessment, and this occurred across development phases. This is an interesting observation, given that although antibodies directed against oligonucleotide drugs have been reported in clinical trials with therapeutic oligonucleotides [13,14], those have generally been deemed of no or negligible impact on pharmacokinetics, pharmacodynamics, or safety [15–20]. The survey data suggest that there may be heightened concerns among HAs with regard to ADAs directed against therapeutic oligonucleotides, although the reasons for this cannot be inferred from the responses.
As part of the survey, we tried to gain some insight into how different companies were approaching the design of nonclinical safety packages to support the clinical development of therapeutic oligonucleotides. So far, HAs have regulated oligonucleotides as small molecules, and it appears that an ICH M3(R2)-aligned approach (ie, two-species toxicology packages) is still very much the default paradigm, with no obvious subclass-related trends. Approximately one-quarter of the respondents had generated single-species toxicology packages, although this was based on project-specific scientific rationale (eg, lack of pharmacological relevance in rodents or low platform/off-target toxicity being expected, due to limited systemic exposure via the route of administration being used). To some extent this aligns to the spirit of approaches outlined in ICH S6(R1). Evidence from the case study responses indicates that HAs were broadly receptive to single-species packages with adequate justification. An interesting observation was a clear split in adopting this more streamlined approach based on company size, with only companies self-identifying as “biotech” or “small-mid pharma” exploring the use of single-species toxicology, invariably favoring the nonrodent. It is tempting to speculate that this could be due to the smaller companies being more willing to take a differential approach to development, versus larger pharmaceutical companies. Certainly, the smaller companies are more likely to be very focused on one specific oligonucleotide class and therefore could be better placed to challenge generic development approaches. It is also possible that they could be working within tighter resource constraints. Both factors could serve as a push to adopting more streamlined, but still science-driven, approaches to enable at least early clinical trials.
Where the two-species toxicology approach was being used, the selection of rodent species was dependent on oligonucleotide class. Companies working on double-stranded oligonucleotides more often reported selecting the rat, whereas the mouse was more frequently the default for single-stranded oligonucleotides, although some still used a case-by-case approach. The reason for this divide is not clear from the survey responses, although two respondents did cite reduced test article requirements for the mouse over rat. Thus, pragmatism likely accounts for a proportion of rodent species selection. Another criterion reported by some of the respondents was the continuity with the rodent species used in prior pharmacodynamic studies. The authors believe that yet another plausible explanation is the recognized sensitivity of the rat to single-stranded phosphorothioated oligonucleotides exacerbating age-related chronic progressive nephropathy (CPN), a species-specific phenomenon with limited or no clinical relevance [21,22]. Although there is evidence of both rodents still being used in the carcinogenicity assessment of single-stranded oligonucleotides, the rat studies afford limited or no multiple to clinical exposure due to CPN-driven survivability issues [13,23].
There was some variation in the nonrodent species selection, although over half (52%) of respondents always defaulted to the use of NHPs. This is likely driven by multiple factors, which may include historical data and understanding of the clinical translation of class toxicities and pharmacokinetics defined in NHPs, as well as lower sequence divergence between human and NHPs [meaning they are more likely to be pharmacologically relevant for the primary and to some extent hybridization-mediated OTEs for certain subclasses, ie, siRNA, single-stranded antisense oligonucleotides (ASOs)]. However, 45% of the companies did indicate that alternative nonrodents had been used to support their oligonucleotide projects, including minipig, dog, and rabbit, where the latter was reported as being used for continuity with the nonclinical disease model. The use of NHPs is currently both constrained by global sourcing issues and the object of increasing 3R considerations. Interestingly, among prior nonusers of alternative species, only 28% stated that they would keep on exclusively using NHPs as their nonrodent species.
Of note, the survey did not address the opportunities or challenges of using animal-active analogues (also known as surrogate molecules) to explore on-target liabilities. This is definitely a gap as it could have shed a complementary light on the issue of species selection for oligonucleotide projects.
When companies were asked to define how they approached high-dose selection for definitive toxicity studies, responses were surprisingly varied and not always aligned to ICH M3(R2) [3]. Two-thirds of the companies indicated that clinically relevant exposure multiples are targeted. There were also individual examples of a “platform MTD” being used, as well as using species-specific approaches (MTD/MFD in rodents, while using targeted exposure multiples for the nonrodent). Among those favoring exposure multiples, the lower fold factors prevailed (10 × , then 25 × ). Distribution did not markedly differ between single-stranded and double-stranded designs, although the 50-fold was only mentioned for the latter. The survey indicated that these diverse approaches have been accepted by HAs, assumedly with scientific justification. However, there was one example provided where an HA did not agree to setting the high dose based on a 10-fold margin and instead requested a 50-fold margin [consistent with ICH M3(R2)] [3]. Of note is that the Japanese HA, PMDA, has recently issued a draft guideline focused on the nonclinical safety assessment of nucleic acid medicines that recommends selection of the high dose can be based on MTD, saturation of exposure, MFD, or a dose providing 50-fold higher exposure than clinical dose [24,25], which is aligned to ICH M3(R2).
There was some variation in how different companies tackled the problem of defining clinical safety margins based on scaling of dose levels from nonclinical species. This was greatest in the approaches adopted for rodent; however, most companies (75%) were in agreement that 1:1 scaling from NHP to human was valid, irrespective of modality, very much in line with the prevailing view in the industry [26–28]. Based on the survey results and accompanying case studies, there is no evidence that HAs in general have a preferred scaling approach, since there were no respondents indicating that their methodology was not accepted. The authors conclude that while multiple approaches may be acceptable, having alignment among regulators and sponsors on the most appropriate scaling methodology would be advantageous in setting consistent expectations globally. Ultimately, dose scaling should be driven by exposure data, which may differ as a function of subclasses [28]. The survey data did not enable verification of whether scaling practices were always data-driven or reflected a standard default approach for each respondent.
There is certainly evidence of continuing interest in human OTEs from HAs. The topic was listed in the “top five regulatory challenges” responses in this survey. It is also noteworthy that the FDA's draft guidance on developing therapeutics to treat the chronic hepatitis B virus infection [29] flags the need to consider human genetic diseases for each OTE predicted in silico for ASO and siRNA therapeutics. The management of human hybridization-mediated OTEs of oligonucleotides has been an evolving field, with multiple recent publications [30–36] offering deeper mechanistic insights. In addition, with advances in bioinformatics and next-generation sequencing (NGS) (ie, RNAseq), new potential approaches to both predicting and detecting human-specific OTEs have opened up. Free-text responses to the question dealing with this area indicated that the vast majority of companies still adopt an approach very much aligned to that described as “best practice” in the OSWG 2012 publication [4]. Interestingly, a small number (3/22) of respondents indicated that they are now routinely applying RNAseq-like approaches to evaluate OTEs. Since the conduct of the survey, the Japanese HA has issued an oligonucleotide guidance document, recommending carrying out in silico and/or in vitro OTE evaluation in a human system [24,25]. The authors anticipate that this trend will continue, as more companies start to leverage the power of NGS. Much work remains to be done, however, in demonstrating the translational value of in vitro transcriptomics. To the authors' knowledge, there are so far very limited data or experience supporting the potential clinical safety significance of OTEs identified using these approaches.
The final section of the survey dealt with perceived relevance of regulatory guidance documents in some detail, with companies initially being asked to score the documents with relevance to oligonucleotide development on a scale of 0–100. ICH M3(R2) [3] and the FDA Center for Drug Evaluation and Research (CDER) Maximum Safe Starting Dose guidance documents [37] ranked most highly, both having mean scores of >70%. All other guidelines had mean scores of ∼50% or lower, with greater variability in responses (reaching as low as zero). Surprisingly, these included the guidance for biologic products, with a significant trend for companies who viewed themselves as “very experienced” in the development of oligonucleotide drugs placing less value on ICH S6(R1) versus those self-reporting as “less experienced.” A notable theme that came out of this section of the survey was the lack of perceived relevance of ICH S2(R1) [8] (genotoxicity testing). These focused predominantly on questioning the need to actually run the standard battery of assays where lack of genotoxicity has previously been defined for well-established chemistries. Based on the OSWG's Genotoxicity Subcommittee analysis published in 2016, there is no evidence of any therapeutic oligonucleotide of established chemistry and subclass testing positive in the standard battery of genotoxicity assay [5]. Based on this, waiving of the standard battery of genotoxicity assays does have significant merit where historical data indicate the risk has been adequately discharged for a given chemistry. Of note, several respondents reported having successfully submitted packages without genotoxicity studies or readouts although the survey data do not enable confirmation of how many of those cases were in noncancer indications.
The management of impurities and their qualification was another area of focus in this section, related specifically to the ICH Q3 guidance documents [9,10]. Although the average perceived relevance score was relatively high (61%) for these, nearly two-thirds (14/22) of companies indicated they intended to apply an impurity threshold of 1.5% for product-related impurities, that is, 10-fold higher than the small-molecule default threshold as defined in these documents. It was interesting to note that to date, no company (at the time of the survey, ie, shortly after the issuance of the Capaldi et al. recommendation [11]) reported having taken the decision to apply the threshold proposed, with case study examples implying that sponsors are currently defaulting to qualification of impurities. However, most respondents (15/22) indicated support for the drafting of oligonucleotide-specific guidance on the management of impurities, suggesting that industry may favor such an approach. Since the survey was conducted, a joint industry/government WG in Japan has reviewed the Capaldi et al. proposal in a position article [38]: consensus was reached around the principle of ultimately setting oligonucleotide-specific thresholds higher than those used for small molecules; however, the number of clinical programs to date was deemed insufficient at this stage to establish such general guidance. Eventually, the WG defaulted to a case-by-case approach, where the principle of toxicological coverage of the impurities should prevail.
When respondents were asked for their view on whether regulatory guidance is required that better addresses oligonucleotide development issues, at least 50% favored this, with only 1/22 responding that the status quo of no formal guidelines was preferred. The free-text responses offered some insight into the rationale for these responses (Table 13) and the survey indicates that ICH M3(R2), management of impurities, and relevance of genotoxicity testing should be prioritized by HAs and industry. The authors of the survey reported on our findings during a session at the DIA/FDA Oligonucleotide-Based Therapeutics Conference, Bethesda, in October 2019, as part of a session on regulatory guidance, and their relevance to oligonucleotides. The presentations were followed by a robust debate, with no absolute consensus, as perhaps represented in this survey too. Concerns were voiced about formal guidelines taking a significant time to develop, which is not ideal in the fast-developing field of therapeutic oligonucleotides, the risk being that they could quickly become obsolete or be too prescriptive. Senior Japanese delegates were promoting the creation of an oligonucleotide-specific nonclinical safety guideline, which has since then progressed in Japan, in the form of a document laying out general principles and referring to existing small- and large-molecule guidance, where relevant [24]. Beyond the issue of official guidelines, a number of points were forcefully made in relation to the value that white papers addressing some of the more unique aspects of oligonucleotides could bring (eg, on managing OTEs or impurities). There was broad agreement on the value of groups such as the OSWG or the EFPIA Oligonucleotide WG. Representatives from HAs in the room encouraged both the WGs and individual players to continue to publish, sharing data and best practice approaches, such as the many white papers that have already been published by the OSWG and others [4,5,39–42].
In conclusion, the EFPIA Oligonucleotide WG survey conducted in 2018 delineated trends in nonclinical safety practices and the regulatory view on this growing field. This included documenting inconsistencies in the industry, the extent of which was maybe unexpected. These data advocate for harmonization efforts whether it be through best practice articles or through guidance. With this modality having now reached a steep slope of expansion, further active collaboration among sponsors and regulators would be an invaluable opportunity to upgrade oligonucleotide development globally.
Footnotes
Acknowledgments
EFPIA Secretariat (David Fernández and Pär Tellman) for executing survey administration as a third party and the Roche EFPIA liaison Melanie Guérard. Pascal Kuner, Roche pRED Datascience, for conducting the retrospective portfolio search. Julia Toth, Tanja Bayer, and Hélène Pierre, Roche pRED Pharmaceutical Sciences, for their kind review of the text, tables, and figures. Former EFPIA oligonucleotide WG members: Kurt Black (Amgen) and Fred Selan (Janssen). Key Opinion Leaders who supported the project: Art Levin (Avidity Biopharma), Scott Henry (Ionis Pharmaceuticals), Takahiro Nakazawa (ANGES), Yoko Hirabayashi (NIHS), Cathaline den Besten, Lola Tome, and Imke Veltman (ProQR Therapeutics). The 22 respondents who took the time and effort!
Author Disclosure Statement
The authors declare no conflict of interest given the precompetitive nature of this work and their respective employers at the time of publication are detailed in affiliations.
Funding Information
No funding was received for this work.