
Abstract
RNA interference (RNAi) applications have evolved from experimental tools to study gene function to the development of a novel class of gene-silencing therapeutics. Despite decades of research, it was not until August 2018 that the US FDA approved the first-ever RNAi drug, marking a new era for RNAi therapeutics. Although there are many limitations associated with the inherent structure of RNA, delivery to target cells and tissues remains the most challenging. RNAs are unable to diffuse across cellular membranes due to their large size and polyanionic backbone and, therefore, require a delivery vector. RNAi molecules can be conjugated to a targeting ligand or packaged into a delivery vehicle. Alnylam has used both strategies in their FDA-approved formulations to achieve efficient delivery to the liver. To harness the full potential of RNAi therapeutics, however, we must be able to target additional cells and tissues. One promising target is the folate receptor α, which is overexpressed in a variety of tumors despite having limited expression and distribution in normal tissues. Folate can be conjugated directly to the RNAi molecule or used to functionalize delivery vehicles. In this review, we compare both delivery strategies and discuss the current state of research in the area of folate-mediated delivery of RNAi molecules.
Introduction
RNA interference (RNAi) is a natural regulatory mechanism that uses small noncoding RNA molecules to inhibit translation [1]. The endogenous triggers of RNAi include short interfering RNAs (siRNAs) and microRNAs (miRNAs). Although these molecules share many similarities, they have distinct modes of action. Long, double-stranded RNA is cleaved by Dicer into ∼21–23 nucleotide siRNAs, with 3′ overhangs, which are then incorporated into the RNA-induced silencing complex (RISC) [2,3]. In the latent complex, the siRNA duplex is unwound by Argonaute 2 (Ago2), and the sense strand is cleaved between base pairs 9 and 10 relative to the sense strand 5′ end [4]. The antisense strand remains bound to the now active RISC and is used as a guide sequence to locate and cleave the target mRNA with which it is fully complementary [2].
The gene-silencing mechanism of miRNAs differs from that of siRNAs. Before Dicer processing, the primary miRNA (pri-miRNA) is cleaved by Drosha to form a pre-miRNA, which is then transported to the cytoplasm by Exportin 5 [5]. Dicer processes the pre-miRNA into ∼19–25 nucleotide miRNAs, with 3′ overhangs, which are then loaded into RISC forming a new complex called miRISC [6]. The miRNA duplex is unwound, releasing the sense strand and leaving the antisense strand as the guide sequence. Unlike siRNAs, miRNAs are only partially complementary to the target mRNA and mediate gene silencing through mRNA cleavage and translational repression [7,8]. Synthetic siRNA and miRNA molecules are compatible with the endogenous RNAi machinery and have been investigated as both experimental tools and gene-silencing therapeutics [9–13].
Many diseases are characterized by aberrant gene expression, making RNAi molecules ideal therapeutics. Despite this potential, the development of RNAi therapeutics has been limited by the inherent nature of RNA, which poses challenges like poor cellular uptake, immune activation, and off-target effects [14]. Several chemical modifications have been investigated to mitigate these effects and improve the pharmacokinetic profiles of RNAi molecules. This includes backbone modifications, such as phosphorothioate (PS) and boranophosphate, to increase nuclease stability [15,16] and sugar modifications, like 2′-O-methyl (2′-OMe) and 2′-deoxy-2′-fluoro (2′-F), to increase thermal stability, reduce immune activation, and improve nuclease resistance [17]. Nevertheless, the delivery of RNAi molecules remains a major challenge in the development of RNAi-based therapeutics. Current delivery strategies involve either encapsulation within a delivery vehicle or conjugation to a targeting ligand. This review will discuss both strategies and highlight the use of folate as a tumor-targeting ligand in various clinical applications, with an emphasis on RNAi molecules.
RNAi Delivery Issues
RNAs are unable to cross the hydrophobic cell membrane due to their large size, hydrophilic nature, and polyanionic backbone. The challenges associated with RNAi delivery strategies have been reviewed extensively [18–21]. Nontargeted oligonucleotides tend to accumulate in the liver and kidneys [22], so it is not surprising that the most successful RNAi-based therapeutics have targeted this organ [23]. To exploit the full potential of RNAi therapeutics, however, efficient extrahepatic delivery must be achieved. Other notable limitations include nuclease stability, immunogenicity, and off-target effects, but the rate-limiting step for oligonucleotide delivery is certainly endosomal escape. Regardless of the delivery method used, oligonucleotides are generally internalized by endocytosis. Multiple endocytic pathways have been identified and have been found to result in successful oligonucleotide uptake [24]. Early endosomal vesicles fuse into a late endosome, which is rapidly acidified by the membrane-bound ATPase proton pump [25–27]. Oligonucleotides must be translocated from the late endosome into the cytoplasm. If this does not occur, the late endosome will eventually fuse with lysosomes and be further acidified. Digestive enzymes in the lysosome will promote nucleic acid degradation, preventing RNAi activity. With these limitations in mind, many research efforts are now focused on the development of safe and efficient delivery systems for RNAi molecules.
FDA-Approved RNAi Formulations
In August 2018, Alnylam's ONPATTRO® (Patisiran) became the first RNAi-based drug to receive US FDA approval, marking a new era for RNAi therapeutics. Patisiran treats hereditary transthyretin amyloidosis (hATTR) with polyneuropathy by targeting transthyretin (TTR) [28]. This was followed by the US FDA approval of GIVLAARI® (Givosiran), in November 2019, which treats acute hepatic porphyria by targeting aminolevulinic acid synthase 1 (ALAS1). [29]. Although both siRNA drugs target the liver, they use different delivery strategies (Fig. 1).
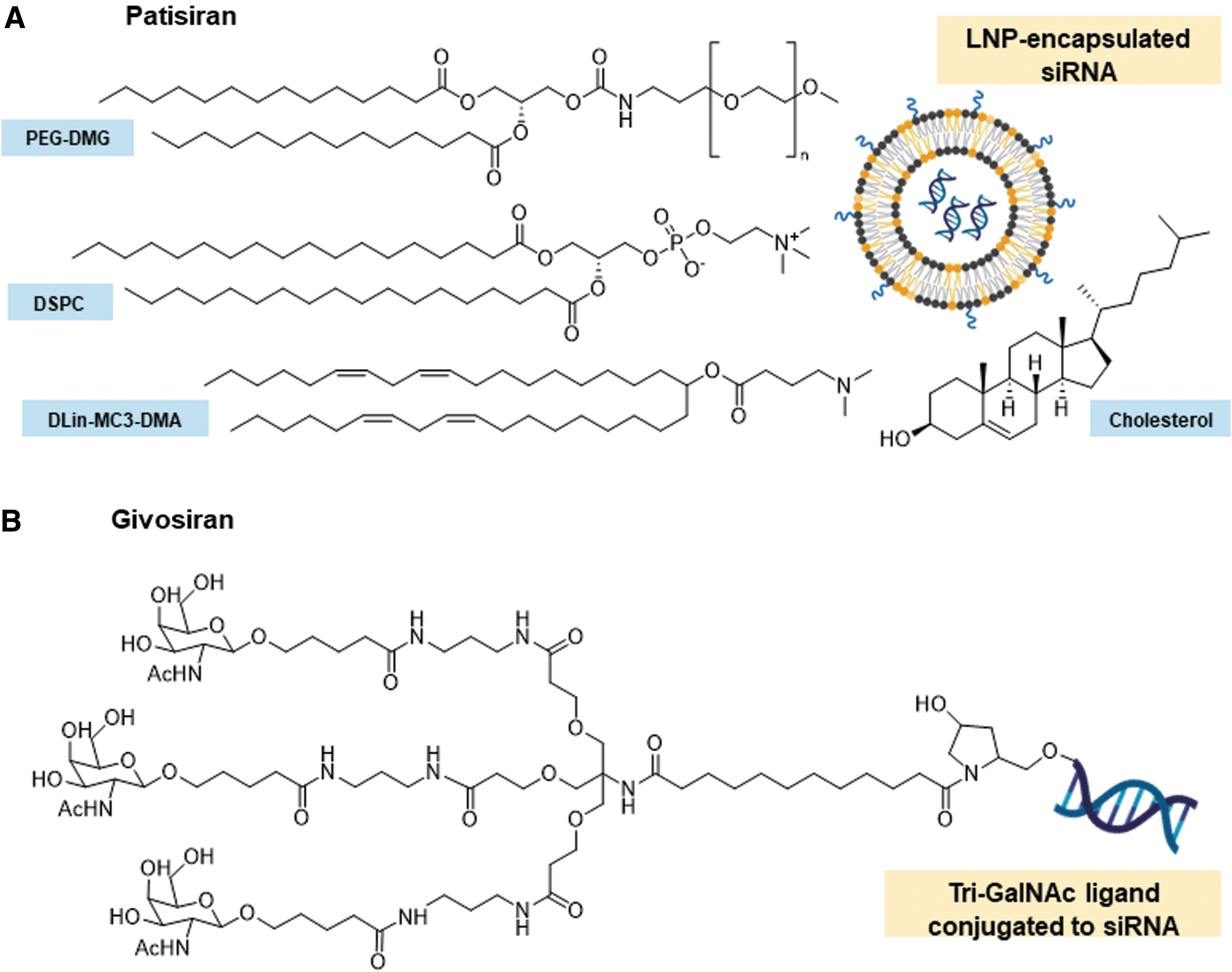
Composition of Patisiran and Givosiran's delivery vehicles/ligands.
Patisiran uses a multicomponent lipid nanoparticle (LNP) formulation and is administered intravenously [28]. LNPs encapsulate the siRNAs, protecting them from enzymatic degradation and shielding their negative charge [30,31]. It has been proposed that LNP uptake in the liver is mediated by apolipoprotein E (ApoE) (Fig. 2) [31,32]. After ApoE associates with the LNP, it facilitates endocytosis through ApoE-binding cell surface receptors, such as the low-density lipoprotein receptor. As the pH of the endosome decreases, the ionizable lipids of the LNP are protonated. These positively charged lipids interact with the negatively charged endosomal lipids, destabilizing the endosomal membrane and causing the disintegration of the LNP [31,32]. This results in the release of the siRNAs into the cytoplasm.
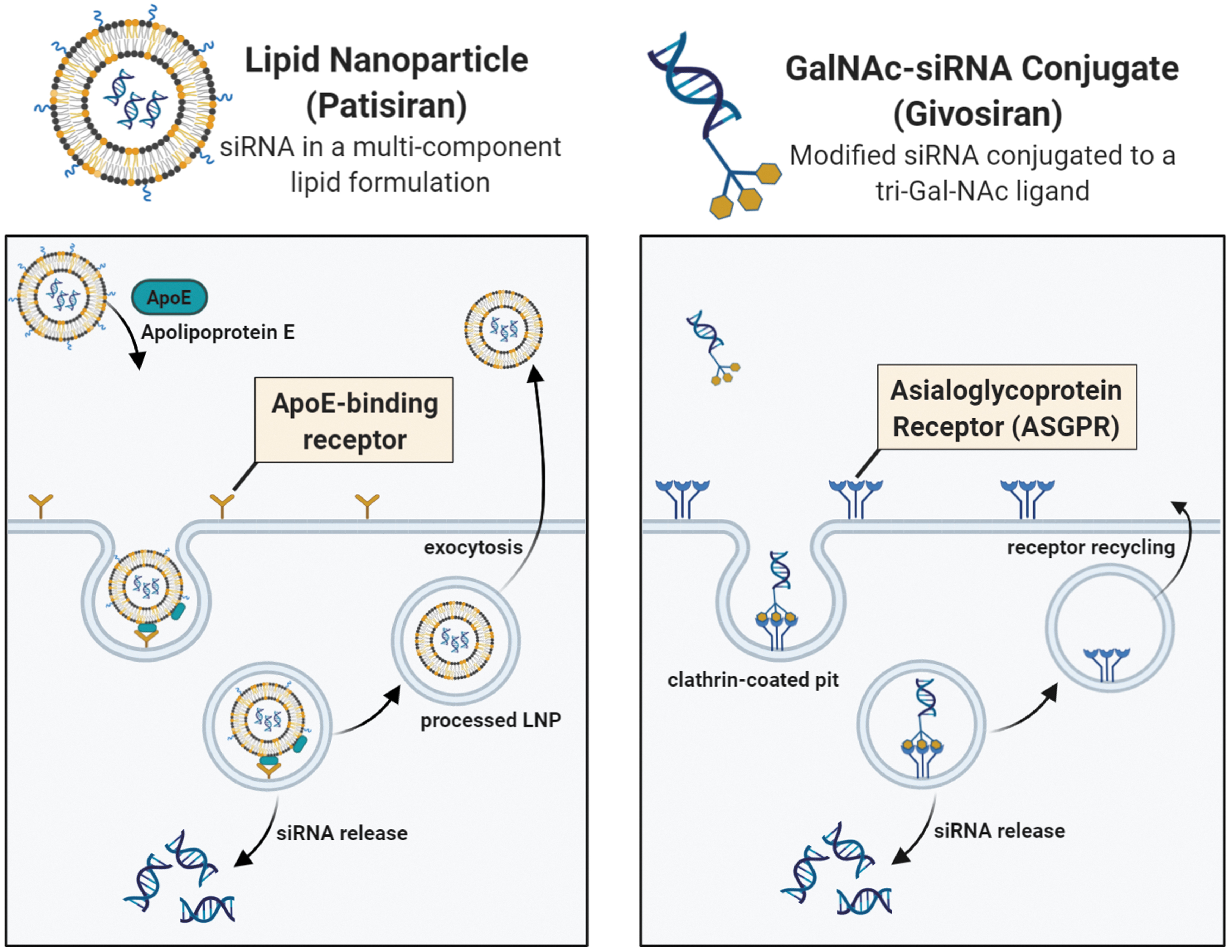
Proposed internalization mechanisms of Patisiran and Givosiran in the liver. Left panel: Uptake of Patisiran's LNP is mediated by ApoE after intravenous administration. ApoE binds to ApoE-binding cell-surface receptors in the liver and aids in internalization of the LNP. Following endosomal acidification, the siRNA is released into the cytoplasm although the majority of the LNPs are recycled back into circulation using exocytosis [31,32]. Right panel: Uptake of Givosiran's GalNAc-siRNA is mediated by the ASGPR after subcutaneous administration. The tri-GalNAc ligand, conjugated to the siRNA, binds the ASGPR leading to receptor-mediated endocytosis. Endosomal acidification results in siRNA release into the cytoplasm and allows the ASGPR to be recycled back to the cell surface [30]. Color images are available online.
Givosiran, on the other hand, is made up of a modified siRNA conjugated to a tri-GalNAc (N-acetylgalactosamine) ligand and is administered subcutaneously. GalNAc binds to the asialoglycoprotein receptor (ASGPR), a cell surface receptor highly expressed in hepatocytes [30,33]. GalNAc-siRNA conjugates are internalized through receptor-mediated endocytosis (Fig. 2). After endosomal acidification, siRNAs are released into the cytoplasm and the GalNAc ligand is rapidly cleaved and degraded, whereas the ASGPR is recycled onto the cell surface [30]. Unlike the LNP formulation, GalNAc-siRNAs are smaller and can be synthesized under solid-phase conditions but also require extensive siRNA modification to provide protection from nucleases [30,34]. Alnylam's GalNAc-siRNAs use Enhanced Stability Chemistry (ESC). They are fully modified using 2′-F and 2′-OMe groups, as well as PS linkages at key positions [35,36].
Alnylam's next generation GalNAc-siRNA conjugates use an ESC+ design, which introduces thermal destabilizing modifications, like glycol nucleic acid (GNA), in the siRNA antisense seed region. The ESC+ design reduces off-target effects and provides enhanced specificity [37]. Despite the success of Patisiran's LNP formulation, Alnylam's current clinical pipeline is focused on exploiting the ESC-GalNAc delivery platform (Table 1). Late-stage development RNAi drugs (Lumasiran, Inclisiran, Vutrisiran, and Fitusiran) use the ESC-GalNAc design, whereas early-stage development RNAi drugs use the ESC+-GalNAc formulation.
Alnylam's RNA Interference Therapeutics Clinical Pipeline
Status as of August 2020.
Approved in the United States and Canada for the treatment of polyneuropathy of hATTR amyloidosis in adults. Approved in the European Union, Switzerland, and Brazil for the treatment of hATTR amyloidosis in adults with stage 1 or 2 polyneuropathy. Approved in Japan for the treatment of TTR-type familial amyloidosis with polyneuropathy.
Approved in the United States and Brazil for the treatment of adults with AHP. Approved in the European Union for the treatment of AHP in adults and adolescents aged 12 and older.
Novartis is leading the development of Inclisiran.
Sanofi Genzyme is leading the development of Fitusiran.
Dicerna is leading and funding the development of ALN-AAT02 and DCR-A1AT and will select which candidate to advance in development.
Developed in collaboration with Vir Biotechnology.
Developed in collaboration with Regeneron.
Developed in collaboration with Vir Biotechnology.
ESC, enhanced stability chemistry; ESC+, enhanced stability chemistry for improved specificity and reduced off-target effects; NDA, new drug application; MAA, marketing authorization application; CTA, clinical trial authorization; IND, investigational new drug.
Bioconjugates for RNAi Molecules
Bioconjugation has been widely used as a delivery strategy for RNAi molecules. Some bioconjugates, like lipids and sterols, can increase cellular uptake through natural transport mechanisms. Cholesterol was the first reported conjugate used for systemic siRNA delivery [45] and has been widely used as a direct carrier for RNAi molecules [46–49] and for LNP functionalization [50]. Although many lipids will accumulate in the liver, extrahepatic delivery can be also achieved with some lipid conjugates [51].
Other bioconjugates target cell surface receptors and can be utilized for selective delivery to target cells and tissues. GalNAc has been the most successful conjugate for this purpose, as its target, the ASGPR, is not only highly expressed in hepatocytes but also has a short recycling time (10–15 min) [52,53]. In addition, GalNAc-conjugate activity is retained even after a 50% reduction in ASGPR expression [54]. Because of this, GalNAc conjugation has become the delivery system of choice for hepatocyte targeting and has paved the way for targeting other tissue types using similar strategies. Herein, we focus on folic acid, the synthetic form of folate, which has been investigated as a targeting ligand for delivery to tumor cells using folate receptors (FRs).
Folate and folate transport
Folates are a group of essential B9 vitamins that play a key role in mammalian one-carbon metabolism. They serve as cofactors in a variety of metabolic reactions and are required for the synthesis of purines, the pyrimidine thymidine, and the amino acids glycine, serine, and methionine [55,56]. Folates are hydrophilic molecules that are polyanionic at physiological pH and therefore cannot readily diffuse through cellular membranes.
Mammals have evolved several systems to transport and uptake folates [57]. The reduced folate carrier (RFC), which is expressed ubiquitously, is the major transport system for folates in mammals and plays a vital role in in vivo folate homeostasis [58]. The RFC relies on a bidirectional anion-exchange mechanism to pump folates into the cytoplasm. It has high affinity for reduced folates but poor affinity for oxidized folic acid [59]. The proton-coupled folate transporter (PCFT) functions at low pH and transports folates using a transmembrane proton gradient. The PCFT is the major transport system in the small intestine and is highly expressed at the apical brush-border membrane of the duodenum and the proximal jejunum where folates are absorbed [60]. In addition to this, the PCFT is involved in folate transport into the central nervous system [61].
Finally, the FR is expressed on the cell surface and transports folates and folate conjugates with high affinity [59]. FRs cluster in invaginations of the cell membrane. Once the folate ligand binds, the receptors are internalized as the membrane transiently closes. Inside the cell, the endosome's acidic environment promotes the release of folate from the receptor and into the cytoplasm, allowing the FR to be recycled onto the cell surface [62–64].
There are four known FR isoforms in humans: FRα, FRβ, FRγ, and FRδ encoded by FOLR1, FOLR2, FOLR3, and FOLR4, respectively. Out of these, the α, β, and δ isoforms are glycosylphosphatidylinositol (GPI)-anchored receptors, whereas the γ isoform is a soluble protein found only in hematopoietic cells [64–66]. FR-δ has been found on ova and regulatory T cells [67]. Notably, FRα and FRβ share ∼70% homology and similar affinities for folic acid, but they have different tissue distribution [68]. FRα is the most widely expressed and studied isoform in humans. This isoform has minimal physiological roles after embryogenesis and thus is expressed at low levels in most nonmalignant tissues. The expression of FRα is restricted to tissues involved in folate resorption or embryonic development, including placenta, kidney, lung, breast, fallopian tubes, and choroid plexus tissues [69–72], whereas FRβ is expressed on activated myeloid cells involved in inflammatory and autoimmune diseases [73,74].
FRα Targeting in Oncology
FRα is highly expressed on numerous cancerous tumors, including 90% of ovarian carcinomas, as well as breast, endometrial, lung, brain, and kidney cancers [71,72]. Due to its low expression in nonmalignant tissues and high affinity for folic acid (Kd < 1 nM), FRα has become an important biomarker in oncology and has been exploited for cancer diagnostics and therapeutics. Folate is a small nonimmunogenic molecule. It is inexpensive and stable over a wide range of pH values and temperatures [75].
Structural and mutational analyses have shown that the pteridine moiety of folate is required for receptor binding, whereas the glutamate moiety is available for conjugation (Fig. 3) [72]. Folate conjugates are recognized and internalized by FRα, making folate a promising ligand for tumor targeting. In 1991, Leamon and Low described the use of folate conjugation to deliver macromolecules through FRs [76], and numerous clinical applications of FRα targeting have been described since (Fig. 4). Table 2 summarizes key FRα-targeted conjugates that have been investigated in clinical trials.
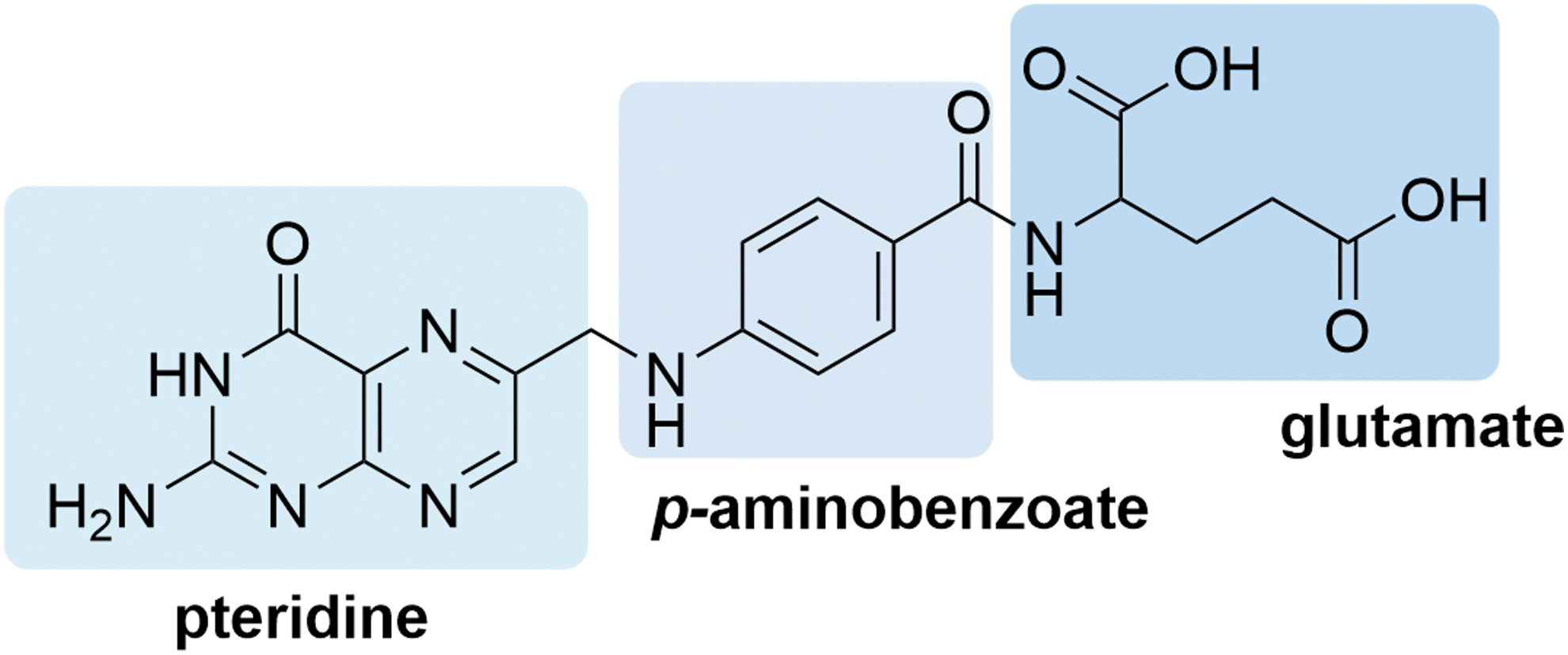
Chemical structure of folate. Folate is composed of a pteridine ring, p-aminobenzoate, and a glutamic acid tail. The pteridine ring is docked deep inside of the folate receptor binding pocket, whereas the glutamate moiety is solvent exposed and is thus available for conjugation. Color images are available online.

Overview of the clinical applications of FRα-targeting in oncology. The FRα is an important cancer biomarker and has been targeted for cancer diagnostics and therapies. Delivery of cytotoxic drugs to cancer cells through FRα can be achieved by conjugation of the drug to a folate ligand to form a folate–drug conjugate. Imaging agents can detect FRα-expressing tumors and serve as companion diagnostics for FRα-targeting therapeutics. Immunotherapy approaches include CAR T cells, vaccines, antibodies, and ADC. Oligonucleotides have been conjugated to folate or encapsulated in folate-functionalized delivery vehicles to target cancer cells. ADC, antibody–drug conjugate; CAR, chimeric antigen receptor; FRα, folate receptor α. Color images are available online.
Key Clinical Folate Receptor α-Targeted Conjugates
NSCLC, nonsmall cell lung cancer; DAVLBH, desacetylvinblastine monohydrazide; TNBC, triple-negative breast cancer.
Immunotherapy
Immunotherapy approaches that target FRα include chimeric antigen receptor (CAR) T cells [77–81] and vaccines [82–84]. Several monoclonal antibodies (mAbs) have also been studied. A notable example is farletuzumab (MORab-003), a fully humanized immunoglobulin G1 (IgG1) antibody that targets FRα. Farletuzumab is thought to induce cell death through various modes of action, including antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, sustained tumor cell autophagy, and inhibition of FOLR1 and Lyn kinase association [85–89]. Farletuzumab has been evaluated in several phase I and II trials [90–94], as well as in a double-blind, randomized phase III study in patients with platinum-sensitive recurrent ovarian cancer [95]. In this phase III trial, the efficacy of farletuzumab in combination with the anticancer drug carboplatin and a taxane was compared to carboplatin/taxane alone but the study's primary progression-free survival (PFS) end point was not met [95]. However, PFS improvements were reported in some patient subgroups after treatment with higher doses of farletuzumab [96].
Another example is MOv18 IgG1, a murine monoclonal antibody, which was generated by vaccinating mice with human ovarian carcinoma cells [97]. A chimeric version of MOv18 IgG1 was later engineered [98], and its safety in patients with ovarian cancer was evaluated in a phase I study [99]. A chimeric IgE antibody (Mov18 IgE) targeting the FRα [100,101], as well as several radiolabeled forms of the MOv18 IgG1 [102–106], has also been investigated.
Anti-FRα antibodies can also be conjugated to cytotoxic drugs to yield antibody–drug conjugates (ADCs) [107,108]. One example is mirvetuximab soravtansine (IMGN853), a conjugate of the maytansinoid DM4, a potent cytotoxic agent, and a FRα-binding monoclonal antibody. The safety and efficacy of this ADC were evaluated in phase I trials either alone [109] or in combination with carboplatin [110]. A randomized, multicenter phase III trial (FORWARD I) compared mirvetuximab soravtansine treatment to other chemotherapeutic drugs (topotecan, paclitaxel, pegylated liposomal doxorubicin) in patients with FRα-positive platinum-resistant epithelial ovarian cancer, primary peritoneal cancer, or fallopian tube cancer [111]. However, this trial did not meet the PFS primary end point in the intention-to-treat and high-FRα populations [112]. A repeat phase III trial is currently recruiting patients with ovarian, primary peritoneal, or fallopian tube cancer whose tumors express a high level of FRα (ClinicalTrials.gov Identifier: NCT04209855).
Imaging agents
FRα-targeted imaging agents have been used as a diagnostic tool to assess the severity of FRα-positive cancers. This includes magnetic resonance contrast agents [113–117], optical imaging agents [118–120], and radioimaging agents [121]. In a phase I/II clinical study, 111In-diethylenetriaminepentaacetic acid (DTPA) folate was evaluated for diagnosis of ovarian malignancy in 35 women [122]. Most patients either had a pathologically proven malignancy or suspected case of new ovarian cancer. 111In-DTPA-folate exhibited rapid target-tissue uptake and nontarget-tissue clearance. However, its use in human imaging was eventually suspended due to high costs in addition to the long half-life of 111In (∼68 h) [121].
Another notable imaging agent is 99mTc-etarfolatide, a peptide derivative of folic acid designed to coordinate 99mTc. This radioisotope is more cost-effective and has a much shorter half-life (∼6 h) than 111In [123]. In preclinical studies, 99mTc-etarfolatide predominantly accumulated in FRα-positive tumor and kidney tissues. It was also found to be removed rapidly from circulation and excreted into the urine [124,125]. Given these findings, 99mTc-etarfolatide has been evaluated in several clinical trials as a companion diagnostic imaging agent to identify tumors that express FRα and that may respond to FRα-targeted therapies [126,127].
More recently, folate has been conjugated to fluorescent dyes for use in image-guided surgery. Van Dam et al. reported real-time intraoperative imaging of ovarian cancer cells using a folate-fluorescein isothiocyanate (FITC) conjugate [128]. Cancer surgery highly relies on visual inspection and palpation to discriminate between malignant and healthy tissues. Folate-FITC (EC17) was able to detect ovarian cancer lesions that were otherwise not detectable by inspection or palpation. However, autofluorescence led to false-positive lesions in ovarian cancer [129]. Infrared dyes, on the contrary, display less autofluorescence and have deeper tissue penetration compared to fluorescein. Pafolacianine sodium (formerly OTL38), another prominent folate-conjugated imaging agent, contains a near-infrared cyanine dye and has been investigated for intraoperative detection of several cancer types, including endometrial [130], nonsmall cell lung cancer [131], and pituitary adenomas [132]. On Target Laboratories, Inc., completed two Phase II clinical trials of pafolacianine sodium for intraoperative imaging of ovarian (ClinicalTrials.gov Identifier: NCT02317705) [133] and lung cancer lesions (ClinicalTrials.gov Identifier: NCT02872701), respectively. In July 2020, On Target Laboratories, Inc., enrolled the first patient in a Phase III clinical trial (ELUCIDATE) to further evaluate the safety and efficacy, as well as the tolerability of pafolacianine sodium, in patients with lung cancer [134].
Folate–drug conjugates
One of the most notable folate–drug conjugates is vintafolide (EC145), a derivative of desacetylvinblastine monohydrazide (DAVLBH) conjugated to folate through a peptide spacer and a disulfide linker. DAVLBH is a vinca alkaloid that inhibits cell division and induces cell death by disrupting the formation of the mitotic spindle [135]. Vintafolide has been tested in various phase I and II trials with promising results [136–138]. However, a phase III randomized controlled trial (PROCEED) was suspended in 2014 due to failure to meet the prespecified outcome of PFS [139]. This trial evaluated the safety and efficacy of the combination of vintafolide and pegylated liposomal doxorubicin. Several other folate–drug conjugates have been studied as well, including EC0489 (a vintafolide analog) [140,141], EC0225 (a vinca alkaloid and mitomycin conjugate linked to folate) [142], and EC1456 (a folate-tubulysin conjugate) [143].
Gene and antisense therapy
Several FR-targeting viral and nonviral vectors for gene therapy have been reported [144–148]. Nonviral vectors include cationic polymers and liposomes [149]. Folate is either linked indirectly through a polyethylene glycol (PEG) spacer or directly to a component of the polymer or lipid [149]. Formulations of FR-targeting polyplexes have used chitosan [150], polyethylenimine (PEI) [151], poly-L-lysine (PLL) [152–154], and combinations of PEG with PEI [151,155] and PLL [153]. In addition, several FR-targeting liposomes have been used in gene therapy [156–160].
This FR-mediated delivery strategy has also been used with antisense oligonucleotides [161–163]. In one study, Lee and Low (1997) reported the use of folate-functionalized liposomes as a delivery vehicle for fluorescently-tagged antisense oligonucleotides, targeting the epidermal growth factor receptor (EGFR), in KB cells (a contaminant of the human cervical cancer cell line HeLa) [164]. The uptake of antisense oligonucleotides encapsulated in the folate liposome was 16-times higher than the control and could be inhibited by addition of 1 mM free folic acid. After 48 h, these antisense oligonucleotides reduced KB cell proliferation by more than 90%.
More recently, Leamon et al. reported the folate liposome-mediated delivery of antisense oligodeoxynucleotides (ODNs) [165]. This formulation was tested in KB xenograft nude mice models after intravenous injection and compared to a nontargeted PEG-containing control. Results show a ∼1.8-fold increase in liver uptake of the folate liposomes compared to the control. However, there was no significant uptake in tumors despite previous reports of in vitro uptake in FR-expressing cells.
Folate-Mediated Delivery of RNAi Molecules
With the success of the receptor-targeting ligand GalNAc and the need for selective extrahepatic RNAi delivery systems, folate has gained a lot of attention as a targeting ligand due to its relevance in oncology. Folate has been mostly used to functionalize delivery vehicles although recent applications have attempted to directly conjugate folate to siRNA and miRNA molecules. This section will highlight key studies in this field and discuss some of the limitations of these delivery approaches.
Packaging siRNAs into folate-functionalized vehicles
Nanoparticle and liposome-based vehicles have been widely used to deliver RNAi molecules into cells. Functionalization of these vehicles with folate for targeted delivery to cancer cells has been reported in several studies with varying results [166–171].
In 2008, Hattori's group reported the synthesis of folate-functionalized LNPs to deliver siRNAs, targeting human EGFR (Her-2), to KB cells [172]. Her-2 is overexpressed in several cancers and is usually associated with poor prognosis [173,174]. Treatment with folate nanocomplexes of anti-Her-2 siRNA decreased cell growth and inhibited the expression of Her-2 in vitro. This delivery system was also investigated in vivo in male BALB/c nu/nu mice bearing KB tumor xenografts. Intratumoral injection of Her-2 siRNA nanoplexes led to reduced tumor growth. This study proposed conditions for the formation of folate-linked nanoplexes, but further in vivo studies are required to assess their safety and efficacy. A comparison between systemic and local delivery would be beneficial, as intratumoral delivery can be impractical for the treatment of certain tumors [175].
As discussed earlier, a major barrier for efficient siRNA delivery is the successful release of siRNA from endosomes. Many strategies have been investigated over the years to promote endosomal escape, such as the use of cationic lipids in lipoplex formulations [176]. Hattori's group recently reported the preparation of cationic liposomes for siRNA delivery [177,178] in addition to three types of folate-PEG liposomes [179]. This last study revealed that the type of cationic lipid used may impact the optimal formulation ratio of folate-poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG2000-DSPE). Notably, formulations with longer PEG chains inhibited cellular uptake of lipoplexes. This is consistent with previous reports that PEG lipids can hinder membrane destabilization, thus decreasing cellular internalization [180].
Once the formulations were optimized, they were tested in KB cells and were able to suppress target enhanced green fluorescent protein (EGFP) and PLK1. Interestingly, intratumoral injections of these folate-PEG lipoplexes in female BALB/c nu/nu mice did not lead to a significant inhibition of tumor growth compared to the control siRNA. This lack of correlation between in vitro and in vivo outcomes suggests that the formulation of these nanoparticle-based delivery systems needs to be optimized and validated for in vivo use, posing a major challenge for the development of folate-functionalized vehicles.
Wagner's group reported similar limitations with their defined folate-PEG siRNA conjugates and polyplexes in 2017 [181]. In this study, a folate-PEG-azide ligand was prepared by solid phase peptide chemistry and was later conjugated to an siRNA bearing an alkyne (hexynyl-ss-C6) at the sense strand 5′-end. KB/eGFP-Luc cells were able to internalize the folate-PEG-siRNAs, and the uptake could be inhibited by the addition of free folic acid. Nevertheless, these siRNAs did not result in gene-silencing activity due to a lack of endosomal escape functionality. To overcome this, the folate-PEG-siRNA was combined with a monodisperse polycationic carrier to build polyplexes. The carrier was composed of three arms, each made up of three succinoyl-tetraethylenepentamine (Stp) units linked by a branching lysine, as well as a cysteine group at each terminus. In this formulation, the proton sponge effect of the carrier's PEI-like 1,2-diaminoethane units can help mediate endosomal escape. The efficacy of these siRNA polyplexes was tested in KB/eGFP-Luc cells. These newly-formulated targeted polyplexes were able to induce significant reporter gene silencing compared to nontargeted and unconjugated controls. This study reports that maximal knockdown was achieved with formulations of 5% folate-PEG-siRNA, highlighting the importance of optimizing formulations for uptake, endosomal escape, and intracellular activity.
Endosomal escape functionality has proven to be an important factor for the formulation of delivery vehicles. However, recent studies suggest that siRNA stability may be equally important. Kataoka and Wagner's groups recently reported the synthesis of targeted siRNA lipopolyplexes, made by coformulations of a PEGylated folate-equipped oligomer, one of three lipo-oligomers, and siRNA [182]. Three formulations were investigated in vitro: TLP1 (a tyrosine-modified oleic acid-based oligomer), TLP2 (a tyrosine-free analog of TLP1), and TLP3 (a linoleic acid-based oligomer).
Translocation profiles of siRNAs from late endosomes to cytosol showed that TLP2 and TLP3, the tyrosine-free formulations, displayed earlier endosomal escape compared to TLP1. All TLPs also displayed similar physiochemical properties. Nevertheless, the intracellular stability of siRNAs in each formulation differed significantly. Despite early endosomal escape functionality, siRNAs in TLP2 and TLP3 were less stable than in TLP1. TLP1 also mediated the best gene-silencing effect and was chosen for further in vivo studies in NMRI nu/nu mice bearing subcutaneous leukemic (L1210) tumors. After intravenous administration, TLP1 downregulated distant FR-directed tumoral EG5 expression by 65% without adverse side effects. The ability of oligonucleotides to escape intracellular endosomes has been a major challenge in the development of effective delivery vehicles. Nevertheless, this study suggests that, for this system, siRNA stability was more significant for gene silencing efficacy than the ability to escape the endosome early.
Altogether, these findings enhance our knowledge and provide a foundation for effective vehicle design. An ideal delivery vehicle should display efficient tissue penetration and cell uptake, provide endosomal escape functionality, be nontoxic and nonimmunogenic, and increase the stability and efficacy of the siRNA and miRNA molecules. Although some progress has been made, there are still some significant limitations associated with some of these delivery vehicles.
Nanoparticle-based delivery is usually limited to clearance organs and requires intravenous administration. The large size of these delivery vehicles can also impede penetration into solid tumors [183], further limiting the tissues that could be targeted. In addition, about 70% of the siRNA internalized by these vehicles will undergo exocytosis [184] and only a small percentage (1–2%) of the total administered siRNA is released into the cytosol [185]. Many cationic polymers and liposomes also display high toxicity in vivo, hindering their clinical applications [186].
Conjugating siRNAs to folate
Direct conjugation of folate or folate derivatives to siRNA and miRNA can help overcome some of the limitations associated with delivery vehicles by improving targeted delivery without the added cytotoxic effects. Unfortunately, the use of folate conjugates in RNAi research has been limited by sophisticated, and often expensive, chemistry. Folate has poor solubility in most organic solvents, apart from DMSO, making it difficult to separate and purify folate conjugates. In addition, direct conjugation to folate usually leads to a mixture of α- and γ-isomers due to the presence of two carboxylic acid groups in the glutamate moiety [187].
Folate phosphoramidites are not commercially available, and there is a lack of reliable protocols for their synthesis. It should be noted that Berry & Associates offered a 5′-folate-triethylene glycol (TEG) cyanoethyl phosphoramidite (BA 0349) in the past (around 2011) at a cost of $843 USD for 100 μmol, but the product has been discontinued and there are no reports using this molecule. Despite this, there have been successful syntheses preparing folate-conjugated RNAi molecules without the use of a folate phosphoramidite (Fig. 5).

Chemical structures of key folate linkers conjugated to siRNA and miRNA molecules.
One of the most well-known attempts to directly conjugate folate to siRNAs was reported by Low's group in 2009 [188]. Folate-conjugated siRNAs were tagged with the fluorophore DY647, and cellular uptake studies were performed in FR-positive RAW264.7 cells. Although these cells were able to internalize folate-siRNA conjugates, further in vitro studies indicated that these siRNAs accumulated in intracellular endosomes.
To test the biodistribution of folate-siRNAs in vivo, nu/nu mice bearing KB tumor xenografts were injected retro-orbitally with DY647-labeled folate-siRNAs. Results showed significantly higher tumor accumulation with folate-conjugated siRNAs compared to the control. This was followed by ex vivo organ imaging, which showed little accumulation of folate siRNAs in healthy tissues (liver, spleen, intestine, muscle, lung, heart, and blood) but high accumulation in the tumor site. This study demonstrated the selective delivery of folate-siRNAs to FR-positive tumors both in vitro and in vivo, showcasing the potential of FR-targeted RNAi molecules as cancer therapeutics. However, it also revealed a lack of endosomal escape functionality, which must be addressed before this system can be fully exploited for targeted delivery to cancer cells.
In 2008, Zhang et al. reported a new strategy to synthesize folate-conjugate siRNAs [189]. In this system, a 17-nucleotide ODN, with a 5′ folate molecule, was used to tether the siRNA through noncovalent interactions. The ODN sequence was randomly chosen and does not code for any known human mRNA. The folate-ODN siRNAs (F-ODN:siRNAs) targeted either αV integrin, which plays an important role in angiogenesis, or survivin, an apoptosis inhibitor.
The gene-silencing activity of these F-ODN:siRNAs was assessed in vitro in the FR-expressing cell lines human umbilical vein endothelial cells (HUVECs) and KB. F-ODN:αV siRNA treatment resulted in ∼80% inhibition of αV mRNA expression but no inhibition of the nontargeted control. In addition, gene-silencing activity of the survivin siRNA (F-ODN:Sur siRNA) was observed in both cell lines, but to a much lower extent in HUVECs. Since siRNA treatment can lead to immune activation, the expression of interferon-β (IFN-β) in HUVECs was measured, resulting in no significant increase in IFN-β mRNA expression after treatment. Overall, these F-ODN:siRNA complexes led to specific cellular uptake and silencing activity in vitro. There are many advantages to this synthetic approach. The preparation of these molecules is simple and cost-effective preparation, allowing for large-scale production. In addition, a single conjugated ODN can simultaneously deliver multiple siRNAs to the target tissue, and ODNs and siRNAs can also be modified to enhance their pharmacokinetic profiles. Despite the potential of ODN:siRNAs, in vivo studies are required to assess their safety and efficacy for future clinical application.
A more popular approach to prepare folate-siRNA conjugates relies on a Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction. In 2012, Carell's group described the synthesis of siRNA conjugated to various derivatives, including folate, using CuAAC click chemistry [190]. This study reported the elaborate synthesis of an azidofolate derivative, from a protected glutamic acid derivative, and an amino-azide tetraethyleneglycol derivative, which was then reacted with a 3′ alkyne-modified oligonucleotide in solution.
Cellular uptake of the resulting folate-siRNAs, bearing a fluorescein label on the antisense strand, was assessed in FR-expressing HeLa cells using confocal microscopy. As expected, folate-modified siRNAs were readily taken up by HeLa cells, whereas the unmodified siRNA controls were not. Folate-siRNAs induced dose-dependent knockdown of target Renilla luciferase in HeLa cells although only 50% knockdown was reported after 1 μM siRNA treatment, the highest concentration tested. Nevertheless, this click chemistry synthetic approach could be further used and optimized for the preparation of folate-siRNA conjugates, which are notoriously challenging to access.
In 2020, the Desaulniers group reported a simpler CuAAC approach to prepare folate-siRNA conjugates [191]. The azidofolate derivative used in this study was synthesized and purified in a single step by conjugating folic acid to 2-azidoethanamine. A propargyl phosphoramidite was synthesized over three steps and was used for solid phase oligonucleotide synthesis. The propargyl modification was then incorporated at a different position within the sense strand, including the central region. Some reports show that thermal destabilization of the siRNA central region can lead to increase in silencing activity [192], yet this region had not been modified with folic acid before this study. Propargyl-modified oligonucleotides were conjugated to the azidofolate derivative through a CuAAC reaction in solution, and the resulting folate siRNAs were tested in vitro in FR-positive HeLa cells and FR-negative HT-29 cells.
Notably, centrally-modified siRNAs displayed enhanced gene-silencing potency in HeLa cells (∼80% knockdown after 0.75 μM treatment) compared to their 3′-modified counterparts (∼40–65% knockdown after 0.75 μM treatment). Further studies targeted the endogenous gene Bcl-2, an antiapoptotic gene overexpressed in ∼70% of cancers. HeLa cells, which express Bcl-2, were treated with a centrally modified folate siRNA, resulting in ∼70% Bcl-2 knockdown after 1 μM treatment. Overall, this study showed that potent siRNA activity can be achieved in vitro with centrally modified folate-siRNAs, providing a novel way to boost gene silencing activity for further RNAi applications although the results need to be validated in in vivo studies.
In addition to siRNAs, the synthesis of folate-miRNA conjugates (called FolamiRs) was recently reported by the Kasinski group [193]. Folate-ethylenediamine (folate-EDA) was prepared in a peptide synthesis vessel and was used for the synthesis of folate-dibenzocyclooctyne (folate-DBCO) conjugates, either bearing or lacking a reducible disulfide linkage (SS), which were then conjugated to the sense strand 5′ end. In FR-positive MDA-MB-231 (breast cancer) cells, both FolamiR-34a and FolamiR-SS-34a were able to reduce Renilla activity. FolamiR-34a was more stable than its unconjugated counterpart, miR-34a, suggesting that the folate conjugate could provide some protection against serum nucleases.
This study also investigated the activity of FolamiR, tagged with a near infrared dye (NIR), in vivo using animals with MB-231 sensor cell xenografts. A single dose of each NIR-FolamiR (with a releasable or unreleasable linker) was delivered through tail vein injection. NIR-FolamiRs were mostly retained in tumors and cleared from other tissues. However, only the unreleasable NIR-FolamiR was able to induce gene silencing. One explanation for this is that the releasable formulation was found to be highly unstable in serum, whereas the unreleasable formulation was stable for over 6 h.
Another in vivo study tested the specificity of FolamiR-34a in nude mice with FR-expressing cells engrafted on the right shoulder and FR-deficient cells engrafted on the left shoulder. After intravenous administration, FolamiRs accumulated in the FR-positive tumor. Further studies revealed that FolamiR-34a treatment could reduce tumor growth in MB-231 xenograft animals without evidence of whole-organ toxicity or immune activation. FolamiR treatment was effective in an immunocompetent aggressive mouse model. In addition to this, a folate-conjugated siRNA (siLuc2) was able to reduce target firefly luciferase activity in MDA-MB-231 cells, demonstrating that this system is applicable to other small RNAs.
Recently, Kasinski and Low reported a novel strategy to promote endosomal release of folate-RNA conjugates, including FolamiRs, using nigericin [194]. This strategy exploits the difference in solute concentration between the cytoplasm, which is rich in potassium ions, and the early endosome, which is rich in sodium ions. Upon internalization, nigericin gets cleaved from the folate carrier. It then localizes to the endosomal membrane where it can exchange potassium and water for an osmotically inactive proton without compensatory sodium release. This causes an osmotic differential that then leads to endosomal swelling and bursting, facilitating the escape of miRNA and siRNA molecules into the cytoplasm.
The evidence presented in this study shows that this nigericin-folate delivery system facilitates endosomal escape, increases RNA availability in the cytoplasm, and improves RNAi activity. Nigericin is nontoxic at the tested doses and is simple to conjugate to small RNAs, making it a good candidate for clinical applications. Although this system needs to be validated in vivo, it offers a promising solution to the endosomal entrapment challenge that could finally enable the shift of FR-targeted RNAi therapeutics from bench to clinic.
Summary and Conclusions
The lack of safe and effective delivery systems for RNAi molecules has been a major challenge in the development of RNAi therapeutics. After decades of research, GalNAc-conjugated siRNAs have emerged as a simple solution to this delivery issue. GalNAc targets the ASGPR, which is expressed at high levels in hepatocytes, allowing for selective delivery to the liver. While the rapid turnover and recycling time of ASGPRs have contributed to the success of GalNAc siRNAs, the lessons learned with this receptor-targeting approach could be applicable for the development of extrahepatic delivery systems.
Folate has gained a lot of interest due to its high affinity for the FRα, an important biomarker in oncology, and has been investigated for the delivery of RNAi molecules to FRα-expressing tumors. There are two main FR-targeting approaches. The most common approach involves packaging siRNAs into folate-functionalized vehicles such as liposomes and polyplexes. Although there have been some promising results from in vitro and in vivo studies, delivery vehicles can impede uptake into solid tumors due to their large size. In addition, delivery vehicles often display high toxicity in vivo, which can limit their clinical applications.
Another approach involves the direct conjugation of folate to RNAi molecules to avoid the negative side effects associated with delivery vehicles. Surprisingly, despite the great potential of folate-conjugated RNAi molecules, only a few studies have been reported. Part of the limitation is attributed to the sophisticated and often expensive chemistry required for their synthesis and the lack of reliable methods to prepare folate phosphoramidites.
More recently, synthetic approaches based on click chemistry have been used to prepare folate-conjugated siRNA and miRNA molecules with great success in various in vitro and in vivo studies. However, several in vivo studies have reported siRNA and miRNA entrapment in intracellular endosomes. As discussed earlier, endosomal entrapment is the rate-limiting step when it comes to the delivery of RNAi molecules yet there are few research efforts focused on the development of folate delivery vehicles with endosomal escape functionality. More resources need to be put into the development of simple and cost-effective folate conjugates for RNAi research to allow for further investigation of these in vivo effects, as well as the delivery mechanism. If the synthesis of folate conjugates can be streamlined and the endosomal entrapment challenge can be addressed, these types of molecules have the potential of contributing to the next generation of RNAi therapeutics.
Footnotes
Acknowledgment
Illustrations were created with Biorender.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.