
Abstract
Nucleic acid aptamers are innovative and promising candidates to block the hallmark event in the prion diseases, that is the conversion of prion protein (PrP) into an abnormal form; however, they need chemical modifications for effective therapeutic activity. This communication reports on the development and biophysical characterization of a small library of chemically modified G-quadruplex-forming aptamers targeting the cellular PrP and the evaluation of their anti-prion activity. The results show the possibility of enhancing anti-prion aptamer properties through straightforward modifications.
Introduction
The prion protein (PrP) is involved in rare, but fatal neurodegenerative disorders affecting humans and animals, known as transmissible spongiform encephalopathies (TSEs) or prion diseases [1,2]. PrP has two alternative forms: a normal, physiologically present, cellular form (PrPC) and a pathologic conformer (PrPSc) [1–3]. PrPC is a soluble protease-sensitive α-helix-rich isoform, while PrPSc is an insoluble protease-resistant β-sheet-rich isoform, whose structure is not known.
The pathogenesis of TSEs is linked to the conversion of PrPC, mainly expressed in the brain, into PrPSc, which has a strong tendency to aggregate, inducing extensive and progressive brain degeneration. Since this conversion is the main triggering event in the development of prion diseases, its inhibition is an ideal target for a therapeutic strategy [4,5]. In principle, tight-binding PrPC ligands are expected to stabilize this form and thus impede its conversion to PrPSc. Interestingly, it has been shown that some PrPC-targeting aptamers successfully reduce the amount of PrPSc in cells persistently infected with the TSE agent [6,7], thus suggesting their potential use in the therapy of prion diseases.
Aptamers are small artificial oligonucleotides that, thanks to their distinctive three-dimensional shape, can bind with high affinity to specific targets and, in most cases, modulate their biological functions [8]. Compared to antibodies, aptamers exhibit significant advantages in terms of size, nonimmunogenicity, and physical and thermal stability. In addition, such nucleic acid-based molecules generally show little or no toxicity and can be efficiently produced by well-established solid-phase synthesis protocols, also allowing the introduction of a variety of chemical modifications ensuring improved bioactivity [9]. Noteworthy, some aptamers showing high affinity for PrPC and significantly reducing PrPSc formation share common sequence and structure motifs, that is, contiguous guanine (G) repeats and G-quadruplex (G4) structures [6,7,10,11].
G4s are higher-order DNA and RNA structures composed of stacks of G-tetrads [12], that is, cyclic arrays of four guanine bases connected through Hoogsteen H-bonds and stabilized by metal cations [13,14], exhibiting high structural variability. Earlier studies found that one of the most effective anti-prion aptamers is an RNA oligonucleotide comprising a four-tandem repeat of the r(GGA) sequence, r(GGAGGAGGAGGA) (R12) [7,11]. R12 folds into a unique G4 structure and forms a dimeric architecture in which the two parallel stranded G4 monomers stack in a tail-to-tail manner through π–π interactions of unusual G(:A):G:G(:A):G hexad planes [15]. On the basis of the structure of R12, an RNA aptamer containing two tandem R12 sequences, r(GGAGGAGGAGGAGGAGGAGGAGGA) (R24), was recently developed [16]. R24 has been shown to form a unimolecular G4 structure, which resembles that formed by two R12 molecules, and also exhibited anti-prion activity.
Interestingly, the DNA versions of R12 and R24 aptamers (named D12 and D24, respectively) share several structural features with the G4s formed by their respective RNA counterparts [17,18]. Indeed, D12 forms a homodimer of intramolecular parallel stranded G4s largely stabilized through base-stacking interactions between G(:A):G(:A):G(:A):G heptad planes of the two G4s [17], while D24 forms a similar structure as a monomeric architecture (Fig. 1) [18]. In addition, D12 has also been shown to bind to PrPC [11,19], although its anti-prion activity is less compared with R12 [7].
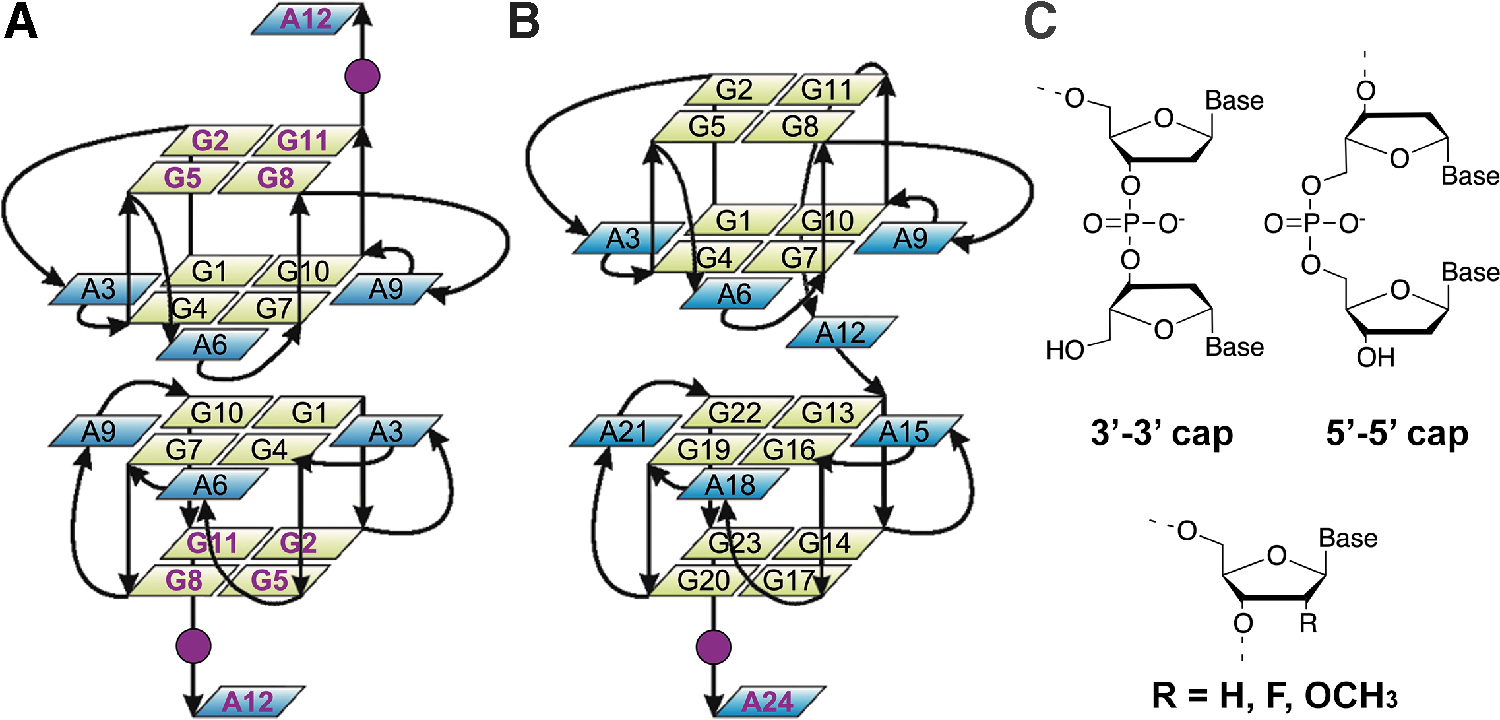
Schematic representation of
Surprisingly, the effects, on such G4-forming DNA, of chemical modifications aimed at enhancing aptamers' resistance against nucleases have never been investigated so far. Indeed, unmodified DNA and RNA aptamers typically undergo rapid enzymatic degradation, confining their use to cases where local delivery is possible [8,9]. For this reason, improving their stability and persistence in biological fluids is a major requirement for an effective therapeutic activity.
In this frame, we report here the rational design, synthesis, and characterization of a small library of chemically modified D12 and D24 derivatives. These modifications include the substitution of the ribose 2′-H with a methoxy group (2′OMe) or a fluorine atom (2′F), and the introduction of oligonucleotide end caps that involve reversing the polarity of the chain (Fig. 1C) [20]. Such modifications have been widely shown to confer resistance toward nuclease degradation and can be readily incorporated into aptamers during solid-phase chemical synthesis [8,9,21].
Materials and Methods
Materials
All common chemicals, reagents, and solvents were purchased from Sigma-Aldrich (Merck group, Italy) unless otherwise stated. Common monomeric building blocks for oligonucleotide synthesis (dA-3′-β-cyanoethyl-5′-dimethoxytrityl phosphoramidite and dG-3′-β-cyanoethyl-5′-dimethoxytrityl phosphoramidite), reagents (amidite diluent, anhydrous CH3CN; deblocking mix, TCA/CH2Cl2; activator, ETT/CH3CN; cap mix A, Ac2O/THF; cap mix B, MI/Py/THF; and oxidizing solution, I2/H2O/Py/THF), and conventional controlled pore glass support (dA-5′-dimethoxytrityl-3′-succinoyl-long chain alkyl amino-CPG, dA-CPG) were purchased from Proligo (Merck group, Germany). Unconventional building blocks (dA-5′-β-cyanoethyl-3′-dimethoxytrityl phosphoramidite, dG-5′-β-cyanoethyl-3′-dimethoxytrityl phosphoramidite, dA-2′-O-Methyl-3'-β-cyanoethyl-5′-dimethoxytrityl phosphoramidite, dG-2′-O-Methyl-3′-β-cyanoethyl-5′-dimethoxytrityl-phosphoramidite, dA-2′-F-3′-β-cyanoethyl-5′-dimethoxytrityl phosphoramidite, dG-2′-F-3′-β-cyanoethyl-5′-dimethoxytrityl phosphoramidite), and the universal CPG support (N-Methyl-succinimido[3,4-b]-7-oxabicyclo[2.2.1]heptane-6-(4,4′-dimethoxytrityloxy)-5′-succinoyl-long-chain alkyl amino-CPG, UniSupport™) were purchased from Glen Research (Maravai LifeSciences). Sensor chips, amino coupling reagents, and buffers for surface plasmon resonance (SPR) measurements were purchased from GE Healthcare. The ovine prion protein (PrPC, A136 R154 Q171 variant, 23–234), kindly provided by Dr. Human Rezaei (Virologie et Immunologie Moléculaires, Institut National de la Recherche Agronomique, France), was produced and purified as previously described [22].
Oligonucleotide synthesis and sample preparation
Oligonucleotides (
Sequences, Apparent Melting Temperatures (T1/2), Unfolding Enthalpy Changes (ΔH), Binding Affinities (Kd), and Anti-Prion Activities of Oligonucleotides
Bold letter = 2′-O-methyl-ribonucleotide; bold and underlined letter = 2′-deoxy-2′-fluoro-ribonucleotide;
Errors were ±1°C.
Errors were within 10%.
Buffer = 100%.
Not determined since the thermal denaturation was not complete at 105°C.
Not determined since cytotoxicity hindered the estimation of anti-prion activity.
Multiple transitions.
CD, circular dichroism; DSC, differential scanning calorimetry; PrP, prion protein; SPR, surface plasmon resonance.
NMR experiments
NMR experiments were carried out using a 700 MHz Varian Unity INOVA spectrometer equipped with a cryoprobe. One-dimensional (1D) 1H NMR spectra of the samples were recorded at 25°C using the pulsed-field gradient DPFGSE for H2O suppression [24]. Data were processed on iMAC running iNMR software (www.inmr.net). Oligonucleotide samples were prepared at 0.2–0.4 mM strand concentration in 0.6 mL (H2O/D2O 9:1) buffer solution containing 10 mM KH2PO4/K2HPO4 (pH 6.8) and 40 mM KCl.
CD experiments
CD experiments were recorded on a Jasco J-815 spectropolarimeter equipped with a Jasco PTC-423S/15 temperature controller. Spectra were recorded in a quartz cuvette with 1 cm path length in the 220–340 nm wavelength range and averaged over three scans. The scan rate was set to 100 nm/min, with 1 s response time and 1 nm bandwidth. Buffer baseline was subtracted from each spectrum. Oligonucleotide samples were prepared at 2 or 4 μM strand concentration (for 24-mer and 12-mer oligonucleotides, respectively) in 10 mM KH2PO4/K2HPO4 buffer (pH 6.8) containing 40 mM KCl. CD melting experiments were carried out in the 25°C–105°C temperature range, at a heating rate of 1°C/min, by following changes of the CD signal at 263 nm, which corresponds to the wavelength of the maximum intensity peak in the CD spectrum of the investigated G4-forming oligonucleotides. Apparent melting temperatures (T1/2) were determined from curve fitting using Origin 7.0 (OriginLab, Northampton, MA).
DSC experiments
DSC measurements were performed using a Nano DSC (TA Instruments). The experiments were performed using an oligonucleotide strand concentration of 0.1–0.3 mM. Samples were prepared in 10 mM KH2PO4/K2HPO4 buffer (pH 6.8) containing 40 mM KCl. Scans were performed at 1°C/min in the 25°C–115°C temperature range. Reversibility was evaluated for each sample by two to three scans after cooling. A buffer–buffer scan was subtracted from the sample–buffer scans, and polynomial baselines were drawn for each scan. Baseline-corrected thermograms were then normalized with respect to the oligonucleotide concentration to obtain the corresponding molar heat capacity curves. The enthalpy change (ΔH) values relative to the dissociation process of oligonucleotide structures were obtained by integrating the area under the heat capacity versus temperature curves [25]. The apparent melting temperatures (T1/2) were estimated as the temperatures corresponding to the maximum of each thermogram peak.
SPR experiments
SPR experiments were performed at 25°C using a Biacore X100 (GE Healthcare) equipped with a research-grade CM5 sensor chip. PrP protein was immobilized using amine-coupling chemistry and HBS-EP as running buffer (HEPES 10 mM, NaCl 150 mM, EDTA 3 mM, and 0.005% Surfactant P20, pH 7.4). The surfaces of flow cells were activated with a 1:1 mixture of 0.1 M NHS (N-hydroxysuccinimide) and 0.1 M EDC (3-(N,N-dimethylamino)propyl-N-ethylcarbodiimide) at a flow rate of 10 μL/min. The protein diluted to a concentration of 10 μg/mL with 10 mM sodium acetate, pH 5.0, was immobilized at a density of ∼300 RU on the sample flow cell, leaving the reference cell as blank. Unreacted activated groups were blocked by injection of 1.0 M ethanolamine at 10 μL/min over the chip surface. Samples were prepared at 32 μM single-strand concentration in 15 mM MES buffer (pH 6.0) containing 70 mM KCl. Kinetic binding data were collected by using the single-cycle kinetics approach [26]. Oligonucleotides were injected sequentially in the same cycle from a low to high concentration (from 0.2 to 3.2 μM) with an association time of 60 s and a long dissociation time (600 s) at the end. Injections were performed at a flow rate of 30 μL/min using 15 mM MES buffer (pH 6.0) containing 70 mM KCl as running solution. No regeneration after each sample was required. The data were fitted to a simple 1:1 interaction model, using the global data analysis option available within the BIAevaluation software provided with the device.
Evaluation of anti-prion activity
The anti-prion activity of oligonucleotides was examined using mouse neuronal cells (GT1-7) persistently infected with the human TSE agent (Fukuoka-1 strain), designated as GT+FK, as described previously [7,27,28]. The cells were grown and maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Equitech-Bio), 50 U/mL penicillin G sodium, and 50 μg/mL streptomycin sulfate (Invitrogen). Approximately 1.5 × 105 cells were plated in each well of a six-well plate, and treatment with each oligonucleotide was started 15 h later. Each oligonucleotide (0.4 mM) was dissolved in a buffer solution containing 10 mM K-phosphate (pH 6.2) and 100 mM KCl, respectively. Fifty microliters of each oligonucleotide solution was added to 2 mL of the medium, the final concentration of each oligonucleotide being 10 μM. As a control, 50 μL of the buffer solution was added to the medium. After 72 h of treatment, cells were lysed in 150 μL of 1 × Triton X-100-deoxycholate lysis buffer (150 mM NaCl, 0.5% Triton X-100, 0.5% sodium deoxycholate, and 50 mM Tris–HCl, pH 7.5), and the supernatant was collected. Samples were digested with 20 μg/mL of proteinase K for 30 min at 37°C. Western blotting for PrPSc was performed as described previously [29]. As the primary antibody, PrP M-20 antibody (Santa Cruz Biotechnology) was used to detect PrPSc. The signals were visualized with Super-Signal (Pierce Biotechnology) and scanned using a LAS-1000 UV mini analyzer (Fuji Film). The density of PrPSc in each solution was measured and compared with that in the control treated with just the buffer solution.
Results and Discussion
Our goal was to design D12 and D24 derivatives with a few numbers of modifications, so that the three-dimensional structure underlying the specific recognition of the target and the thermodynamic stability of G4s would be practically unaffected. With this in mind, the nucleotides involved in the formation of the unusual G(:A):G(:A):G(:A):G heptad planes, which seem to be indispensable for the formation and stabilization of the peculiar architecture of these sequences, were not taken into account for the modifications.
Our quest to improve the aptamer's nuclease resistance thus led to modifications of D12 and D24 by introducing 2′OMe- or 2′F-substitutions in the external G-tetrads (for D12) and/or in the last residue of the sequence (for both D12 and D24), which are the nucleotides most exposed to nuclease attack, and by inserting a 3′-3′ or 5′-5′ inversion of polarity site just before the last adenine residue of the sequences (residues A12 and A24 for D12 and D24, respectively) (Fig. 1). In this way, the polarity of the nucleic acid chain is reversed generating nucleic acids that are barely recognized as substrates by 3′- and 5′-exonucleases. The sequences of the synthesized oligonucleotides are shown in Table 1.
The effects of chemical modifications on G4 formation were first evaluated by monitoring the 1D 1H NMR spectra of the derivatives compared with their natural counterparts (Fig. 2A, B and Supplementary Figs. S1–S4). Validating the G4 folding of modified sequences is important, given that even small changes can have large effects on the structures adopted by G4-forming sequences [30,31]. High similarity between NMR spectra is a strong indication that sequences adopt the same folding topology. The peaks in the imino proton region (10.5–12 ppm) of NMR spectra clearly indicated a single G4 conformation for the two unmodified sequences (Fig. 2A, B) [17,18]. Conversely, some of the modifications inserted led to multiple conformations as observed in NMR spectra. In particular, concerning the oligonucleotides containing a 3′-3′ or 5′-5′ polarity inversion, the emergence of a single major G4 conformation was only observed for the D12 sequence modified with 3′-3′ end cap [D12_3′-3′ (
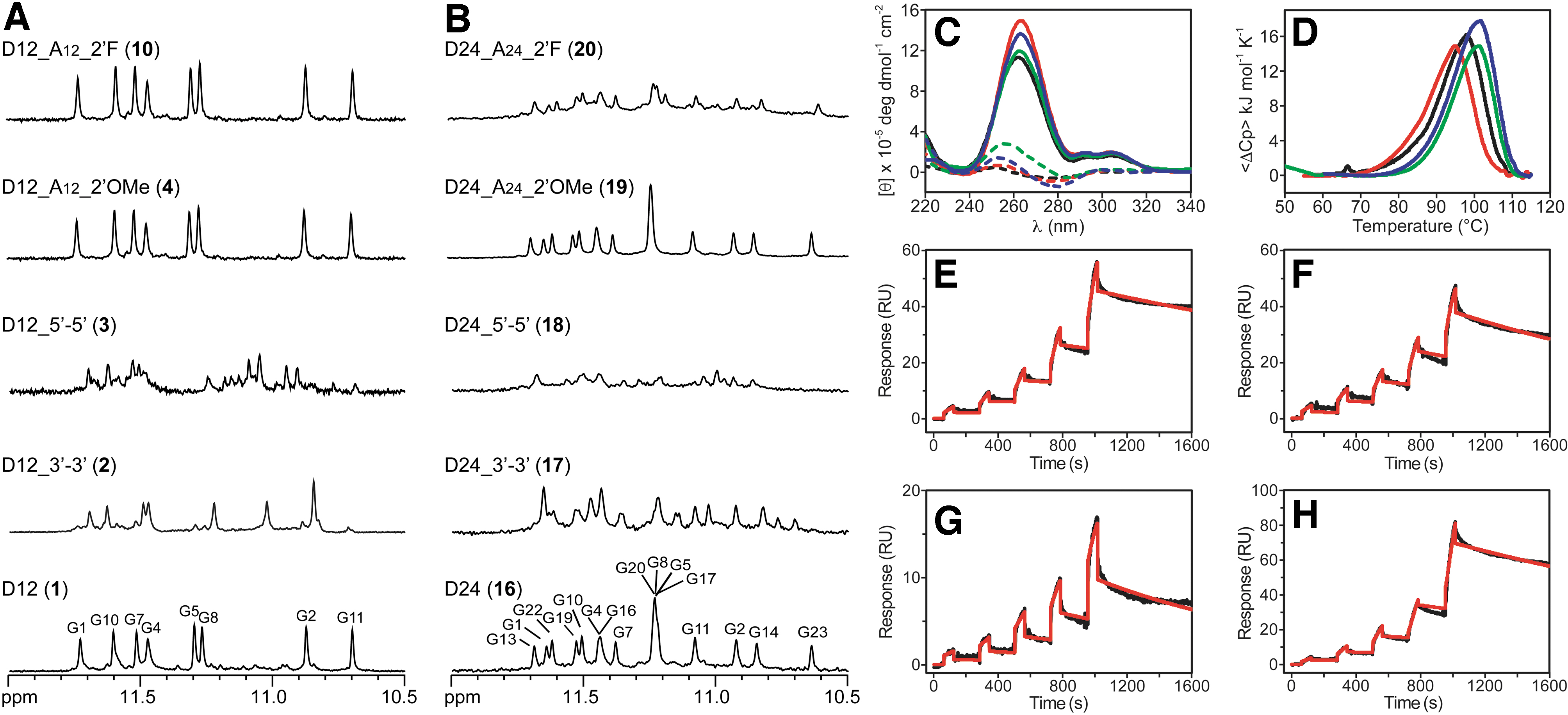
Within the 2′OMe series, the two single-position modified sequences [D12_A12_2′OMe (
Within the 2′F series, the D12 derivatives were all observed to form a single major G4 conformation (Fig. 2A and Supplementary Fig. S3), while the spectrum of the 2′F-modified D24 sequence [D24_A24_2′F (
CD is extremely sensitive to conformational changes of nucleic acids and provides a useful tool for detecting the overall topology of a G4 structure [32]. CD spectra of all chemically modified oligonucleotides in the presence of K+ showed profiles very similar to those of D12 and D24 (
CD melting analysis confirmed the expected high thermal stability of these G4 structures (Table 1 and Supplementary Fig. S6) [17,18]. In particular, the thermal denaturation of most of the 2′OMe-modified D12 analogs was still not complete even at 105°C and did not allow us to estimate the half-point of the thermal transitions in the temperature range analyzed by CD. The other derivatives all showed an apparent melting temperature (T1/2) very close to that of the parent oligonucleotide, except for D12_G2G5G8G11A12_2′F (
However, the thermal profiles for all the oligonucleotides were affected by the kinetics of the process, resulting in a considerable hysteresis between the heating and cooling processes or in irreversible transitions. This was in agreement with the dimeric architecture of D12, but was somewhat unexpected for D24 (because of the fast kinetics usually associated with the formation of unimolecular G4s) and suggests that a fraction of D24 (
As for D12 and its derivatives, the DSC melting curves indicated that they unfold in a monophasic transition. The comparison of T1/2 values derived from DSC (Fig. 2D and Supplementary Fig. S7) confirms, for the modified oligonucleotides, a thermal stability comparable to or higher than the unmodified one, with the only exception of D12_G2G5G8G11A12_2′F (
On the other hand, the DSC thermograms of D24 and D24_A24_2′OMe (
Then, as for all postselection modifications, it was verified that the introduction of such modifications was not detrimental to the aptamers' binding abilities [38,39]. To evaluate the binding properties of modified oligonucleotides to PrPC (Supplementary Fig. S8), we used SPR spectroscopy. SPR has been widely applied to the study of protein–DNA interactions, including G4s, addressing questions about the affinity, kinetics, and specificity of an interaction [38–41].
SPR experiments on the selected D12 analogs, performed employing single-cycle kinetics method, showed a response proportional to oligonucleotide concentration, indicative of a specific interaction with PrPC (Fig. 2E–H and Supplementary Fig. S9). To evaluate the affinity, the obtained binding curves were fitted to a simple 1:1 binding model, giving calculated dissociation constant (Kd) values in the range 45–420 nM (Table 1). The kinetic profile of D12 revealed a Kd value of 70 nM, in agreement with literature data [11,19]. The oligonucleotide modified with 3′-3′ polarity inversion [D12_3′-3′ (
Finally, the anti-prion activity of oligonucleotides
The oligonucleotides described in this communication are the first examples of chemically modified G4-forming anti-prion aptamers. Our findings demonstrated that some modifications aimed at enhancing aptamers' resistance against nucleases can keep their functional folding, stability, and binding properties unaltered (if not improve them) and enhance their anti-prion activity. These results may be used to develop effective next-generation aptamer-based drugs against prion diseases.
Footnotes
Acknowledgments
The authors thank Dr. Iolanda Fotticchia for DSC measurements and Dr. Human Rezaei for generously providing PrPC for SPR studies.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by a Future in Research grant from the Italian Ministry of University and Research (MIUR) to B.P. (no. RBFR13XFXR). M.K. and T.M. were supported by JSPS KAKENHI (nos. 18K19397 and 20H03192, and 16K07269, respectively).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.