
Abstract

Antisense oligonucleotide (ASO) therapeutics are synthetic DNA oligonucleotides, most often containing modified nucleosides, that hybridize to their mRNA targets in a sequence-specific manner and have all or some internucleotide linkages modified as phosphorothioates (PSs). ASOs modulate protein expression via several mechanisms. One of the main mechanisms involves the formation of an ASO–mRNA heteroduplex that elicits RNase H1 activity, mRNA degradation, and downregulation of protein expression as a consequence [1,2]. Other major mechanisms include translational inhibition by steric hindrance and splice modulation.
The idea of introducing a PS linkage by substituting a sulfur atom for one of the nonbridging oxygen atoms in the phosphate group came about in 1967 in Prof. Eckstein's laboratory [3,4]. Ability to synthesize this linkage opened the possibility to compare its properties to phosphodiester linkage [5,6]. It then became evident that this is the most critical modification for
In 1998, the U.S. Food and Drug Administration (FDA) approved a first-generation ASO drug, Fomivirsen (a 21-mer PS-modified DNA analog), for the treatment of cytomegalovirus retinitis in immunocompromised patients [7].
Further to that, several 2′-modifications were subsequently discovered that were associated with favorable pharmacokinetic properties, increased nuclease stability and tissue bioavailability, which led to the approval of second-generation antisense drugs. It appears that over time we gained a better understanding of the mechanism and therefore achieved success in developing ASOs that can activate RNase H1 and reduce gene expression to treat gain-of-function diseases (e.g., Mipomersen for the treatment of homozygous familial hypercholesterolemia [8], Inotersen for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy [9], and Waylivra for the treatment of familial chylomicronemia syndrome [10]; all these drugs are 20-mer PS-modified 5-10-5 MOE [methoxyethyl] DNA gapmers). There has been more focus and therefore success in the development of PS ASOs for treatment of gain of function diseases compared to those for loss-of-function diseases.
The only example so far of approved PS ASO for loss-of-function disease is Nusinersen [11], an 18-mer PS fully 2′-MOE-modified DNA analog that promotes exon 7 inclusion by targeting pre-mRNA splicing and was approved for the treatment of Spinal Muscular Atrophy. This could be due to ASO therapeutics field's deeper understanding of RNase H mechanism relative to splice modulation mechanisms.
While these compelling milestones were achieved in the development of PS ASO therapeutics during the last 30 years, there were a few disappointments such as the FDA rejections of Drisapersen, a 20-mer PS fully 2′-OMe (methoxy) DNA analog intended to restore dystrophin in Duchenne muscular dystrophy (DMD) patients with amenable mutations and Mongersen (NCT02974322), a 21-mer PS DNA analog intended for the treatment of Crohn's disease.
In parallel, several scientists were wondering if properties of PS oligonucleotides would be affected by the absolute configuration of the chiral phosphorus atoms. These considerations were inspired by the success of Sepracor, i.e., the success with Levosalbutamol, the (R)-enantiomer of its prototype drug Salbutamol (a racemic mixture containing both (R)- and (S)-enantiomers), while only (R)-enantiomer is pharmacologically active [12] and the tragic disaster of thalidomide (a racemic mixture containing both (R)- and (S)-enantiomers) where teratogenic effects are induced exclusively by the (S)-enantiomer [13–15]. When one of the nonbridging oxygens in the phosphodiester link is substituted with sulfur to make a PS oligonucleotide, this generates a chiral center, creating Rp and Sp diastereomers from the phosphorus atom (Fig. 1). For a typical 20-mer PS DNA oligonucleotide like Mipomersen, for which the chirality of the phosphorus atom is not controlled during synthesis, the resulting mixture could have up to 219 individual drug molecules [524,288 possible diastereoisomers].
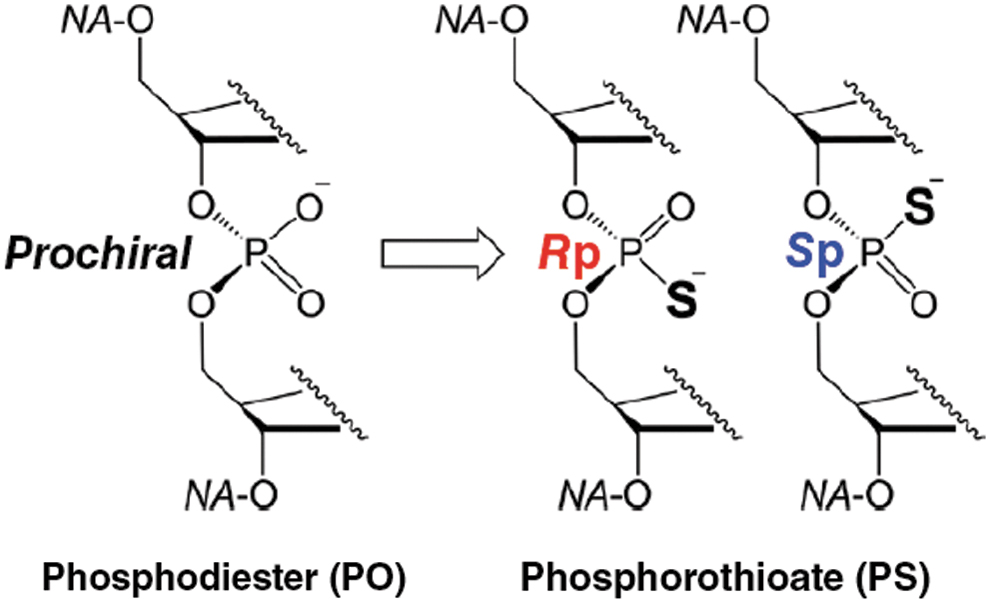
Introduction of chirality in Phosphodiester linkage.
Chemistry
As far back as 1994, Prof. Stec stated, “The effects of the chirality of Oligo-S have so far been unappreciated, because Oligo-S has not yet been synthesized with stereocontrol” [16]. The pioneer work over several years from Prof. Stec's laboratory (oxathiaphospholane method) and Prof. Wada's laboratory (oxazaphospholidine method) ([16,17] at the University of Tokyo and Chiralgen, Ltd. focused on the synthesis of chirally controlled PS oligomers. Dr. Krieg et al. worked to understand the difference in biological response of chirally controlled PS oligomers (the word stereocontrolled is in this article) [18,19] and stereoenriched PS oligomers [20,21].
From those studies, we learned that (1) Sp diastereomer is more resistant to nuclease degradation whereas Rp is more sensitive, (2) Sp diastereomer is more hydrophobic than Rp, (3) Rp modified analogs bind to complementary RNA with slightly higher affinity compared to Sp, (4) Rp CpG analogs show stronger immune activation compared to Sp [18], and (5) RNase H activity varies depending on the stereochemistry of the PS linkage [19]. The challenge of obtaining such Rp/Sp mixed backbone sequences with high stereoselectivity and a chemistry amenable to scaling up the drugs to support animal studies to be able to evaluate the total effect of the above described properties was widely acknowledged. With this intent, Ontorri, Inc., was established in 2009, which later became Wave Life Sciences after a merger with Chiralgen Ltd. Wave used an improved version of the oxazaphospholidine method named SOSICS (stereocontrolled oligonucleotide synthesis with iterative capping and sulfurization) and demonstrated diastereomeric purity of >99% using dimers [22,23].
Recently Published Results Addressing Chirality in PS Linkages
In 2017, Iwamoto et al. from Wave Life Sciences confirmed the impact of Rp/Sp stereochemistry on in vitro metabolic stability, lipophilicity, affinity to the complementary strand, and, most importantly, RNase H activity using a few stereocontrolled versions of Mipomersen [22].
This was the first time that stereocontrol was introduced at all of the PS centers in a 20-mer oligonucleotide, and the effect was studied on lowering of ApoB100 protein in transgenic mice following multiple intraperitoneal injections. Since identifying a few superior diastereoisomers out of 219 possibilities is a challenge, a small set of sequences encompassing diverse physiochemical properties were rationally designed.
To understand the effect of stereochemistry on the DNA gap region and the wing region, all-Rp, all-Sp, Rp-DNA gap/Sp-wings, and Sp-DNA gap/Rp-wings ASO were synthesized. In addition, 2 ASOs were designed with 3′-RpRpSp-5′ and 3′-SpSpRp-5′ blocks in the DNA gap region based on stereochemical preferences predicted from the X-ray structure of RNase H1 bound to an RNA–DNA heteroduplex [24]. Cell-free assays showed higher rate and more selective cleavage of complementary RNA when the rationally designed stereocontrolled ASO/RNA duplex was incubated with a human RNase H1 catalytic domain.
The same sequence in transgenic mice showed statistically significant longer duration of effect with no impact on efficacy compared to the stereorandom control. The authors concluded that the increased effect of RNase H1 activity with the best oligonucleotide compared to the original stereorandom oligonucleotide was attributed to optimal position-specific stereochemistry (3′-SpSpRp-5′) as the distribution of this superior ASO was similar to the stereorandom control in mouse liver. Notably, there was no observed improvement in the potency of the stereocontrolled ASOs over stereorandom control, when tested in cells.
Using a few gene targets, Wave scientists showed that interruption of a continuous stretch of Sp centers by Rp (also represented as-SpSpRpSpSp-) in the DNA gap region impacts RNase H1 selectivity and activity in cell-free assays [25]. The finding that the position of Rp linkage could direct the position of cleavage in the opposite RNA strand led to the principle of allele-specific targeting that was used to develop ASOs; WVE-120101 (PRECISION-HD1) and WVE-120102 (PRECISION-HD2) for Huntington's disease (HD) patients harboring SNP1 (rs362307, T/C) and SNP2 (rs362331, T/C), respectively [26,27]. These candidates are intended to reduce mutant huntingtin (muHTT protein) only while leaving wild-type huntingtin protein wtHTT) intact. As shown in Fig. 2, stereocontrolled ASOs can be designed to target the variant on the mutant allele of heterozygous (carrying different nucleobase at the same position on 2 alleles) SNPs in autosomal dominant disorders like HD where the mutant allele is in linkage disequilibrium with expanded CAG repeats. The feasibility of specifically targeting a nonpathogenic SNP located several thousand bases downstream from the disease-causing mutation in HD has been assessed before by using small interfering RNA (siRNA) [28] or ASOs [29,30]. In addition, allele-specific gene silencing has also been demonstrated in cells and animals by targeting the expanded CAG tract directly for HD and other dominantly inherited neurodegenerative disorders [31,32]. However, none of these modalities have entered the clinic yet.

Stereocontrolled ASO used for Allele specific targeting. Combination of stereocontrolled ASO incorporating “SSR” code with mismatch to selectively cleave mutant allele (fully complementary strand) and leave the wild-type allele with a single mismatch SNP. ASO, antisense oligonucleotide.
Other laboratories have also studied the impact of stereocontrolled ASOs on drug properties [33–36]. The results of these studies where stereorandom and stereocontrolled centers were mixed in the DNA region of ASOs are difficult to interpret and to arrive at a logical conclusion [33,35–37].
In a recent report, Østergaard et al. from IONIS Pharmaceuticals evaluated stereocontrolled PS in 2′-cEt (constrained ethyl) in (3-10-3) 16-mer gapmers targeting CXCL12 mRNA in mouse 3T3–L1 cells and studied caspase activation in Hepa1–6 cells [34]. Oxazaphospholidine monomers were used to prepare the stereocontrolled linkages. Stereocontrolled linkages were introduced in the DNA gap while stereorandom PS centers were present in the wings.
This design was chosen based on previous results that stereochemistry in the wing region did not have an impact on the activity of ASO [36,38]. The sequences containing full Rp and a full Sp gap and then followed by an Sp linkage walked across a full Rp gap and vice versa were synthesized (Fig. 3). With additional data from this report, there is an alignment on the impact of Rp/Sp chemistries on the binding affinity to target and RNase H1 selectivity. The authors concluded that limited use of stereocontrolled PS centers can be used to improve the cleavage of RNA and introduce selectivity. The cleavage maps obtained by Wave and IONIS demonstrated that the position of the Rp center dictates the point of cleavage in RNA (Rp linkage 2 nucleotides downstream from the cleavage site).

Description of Rp or Sp walks.
Furthermore, in both (Wave and IONIS) in-vitro studies, full Sp gap ASOs were less potent and introduction of even a single Rp linkage was sufficient to restore potency relative to the stereorandom parent ASO. In IONIS stereocontrolled designs, incorporation of 1 Rp in Sp backbone led to improved in-vitro potency compared to stereorandom ASO. Introduction of repeat SpSpRp or RpRpSp designs exhibited a potency comparable to the stereorandom ASO; however, the repeat SpSpRp ASO exhibited reduced caspase activation (more toxic) compared to the stereorandom ASO. Notably, IONIS ASOs with stereocontrolled gaps (including the same stereochemistry pattern in the gap as Wave utilized) were not more potent than the stereorandom parent ASO in mice.
Therefore, it was concluded that stereocontrolled ASOs do not provide a significant advantage in vivo and stated that “added cost and complexity of controlling PS chirality makes this a less attractive strategy relative to the gap-modification approach using 2′ and backbone modifications for improving therapeutic profile.” Although there were some variations that were found to be less toxic in in vitro assays and more tolerable in mouse studies, there was not a single stereocontrolled ASO that had improved potency and tolerability.
In another study conducted in Cos7 cells expressing mini ferrochelatase (FECH) gene, stereorandom and stereocontrolled 2′-MOE-modified ASOs were compared for their potency in splice correction [36]. In this study, the Sp-PS isomer was less effective, and the Rp-PS diastereomer was equipotent to the stereorandom ASO.
Discussion and Perspective
Based on the combined results from published works, one could be tempted to dismiss the merit of stereocontrolled oligonucleotides. However, we need to be mindful that the comparison of the results is not straightforward since Wave and others are using different sequences, chemistries, and configurations.
The issue of lower manufacturing yield for stereocontrolled ASOs has been brought up several times by some authors and was also discussed in the Østergaard et al. paper [34]. The % yield and the cost of goods of manufacturing may not be critical issues if there is enough demand and if the process is optimized. In addition, various ways of introducing the stereocontrolled PS center have not been fully explored yet. For example, P(V) building blocks were recently used by Phil Baran's team and collaborators from Bristol Myers Squibb for the incorporation of diastereoselective PS linkage as opposed to traditionally used P(III) building blocks [39]. Although this strategy is far from its use for therapeutic applications, it presents another direction for process optimization. Also, it is worth noting that this P(V) method is already used for manufacturing a cyclic dinucleotide for therapeutic application.
The first oligonucleotide was synthesized using automated synthesizers in the early 80s and if yields had stayed at the same level, no oligonucleotide would be an approved drug today. Back in the 90s, a synthetic oligonucleotide becoming a drug seemed like fiction. Therefore, it could just be a question of time before we see the full potential of stereocontrolled oligonucleotides as therapeutics.
In addition, it has been unambiguously established that minimal introduction of Rp center/s in Sp backbone DNA region helps to direct RNase H1 cleavage of target RNA [25,34]. Utilizing stereocontrolled ASOs for allele-specific targeting is a very convincing rationale and has shown promise in in vitro studies [26,27]. The selectivity of stereocontrolled ASOs to target mutant alleles in patients has yet to be demonstrated as the results of evaluation of WVE-120101 and WVE-120102 that are intended to lower muHTT protein selectively, while keeping wtHTT intact in patients are awaited. If successful, this approach could be utilized for the development of treatments for other autosomal dominant disorders such as HD.
The question we need to answer is the following: what does stereocontrolled design offer that can change the safety and efficacy of the drug? So far, there is no evidence in the literature demonstrating higher bioavailability of stereocontrolled ASOs compared to stereorandom ASOs in vivo. The latest data (although limited) show that Sp and Rp chemistries independently render favorable physiochemical properties to the molecule, but none of the combination/iterations improves potency and safety of the drug.
The very first efficacy data in patients from a stereocontrolled ASO were disappointing, and hence the clinical development of Suvodirsen (a stereocontrolled PS ASO) for treatment of DMD patients with the Exon 51 mutation was discontinued (NCT03907072). Comparison of Suvodirsen with Drisapersen (stereorandom fully 2′-OMe modified PS ASO) showed that in patient-derived myoblasts under free uptake conditions, Suvodirsen restored Dystrophin more efficiently than Drisapersen [40]. It is noteworthy that Suvodirsen chemistry is different from Drisapersen [41]. Drisapersen and Suvodirsen trials were terminated due to lack of efficacy at the maximum dose that could be safely administered to patients, 6 and 4.5 mg/kg, respectively [42,43].
Finally, it would be helpful to the oligonucleotide community to have the opportunity to review the preclinical safety and pharmacology results of Suvodirsen in comparison to Drisapersen. Improved in vitro efficacy is a good sign but is not enough to expect improved efficacy in patients in the absence of other features such as improved bioavailability and/or safety. Access to the preclinical findings of stereocontrolled ASOs may help the scientific community learn and determine the merits of stereocontrolled ASOs over stereorandom mixtures.
Also, it is worth noting that the oligonucleotide field has learned that the combination of a certain SEQUENCE, made out of a certain CHEMISTRY, targeting a certain mRNA, in a certain organ (such as muscle, liver, etc.) or body compartment [such as the central nervous system (CNS), blood, etc.] or a particular cell type for a certain pathology is what makes a drug. Judging the role of stereochemistry from a couple of downfalls may be premature.
Footnotes
Author Disclosure Statement
The statements in this article are authors' opinions and they do not constitute their employers' or clients' opinions. Meena is currently employed at Stoke Therapeutics. M.M.L. discloses being an independent consultant for several companies developing oligonucleotide therapeutics, none of them using stereocontrolled chemistry.
Funding Information
No external funding was received for this work.