
Abstract
Ribosomal protein L3-like (RPL3L) is a poorly characterized ribosomal protein that is exclusively expressed in skeletal and cardiac muscle. RPL3L is also downregulated in Duchenne muscular dystrophy (DMD), suggesting that it may play an important role in muscle biology. In this study, we investigated the role of RPL3L in skeletal muscle of healthy C57 and dystrophic mdx mice. We show that RPL3L is developmentally regulated and that intramuscular adeno-associated virus (AAV)-mediated RPL3L knockdown in the tibialis anterior of C57 and mdx mice results in increased specific force with improved resistance to eccentric contraction induced muscle damage in dystrophic muscles. The mechanism by which RPL3L knockdown improves muscle function remains unclear. Histological observations showed a significant increase in muscle length and decrease in muscle cross-sectional area after RPL3L inhibition suggesting that this ribosomal protein may play a role in myofiber morphology. The endogenous downregulation of RPL3L in DMD may be a protective mechanism that attempts to improve skeletal muscle function and counteract the dystrophic phenotype.
Introduction
Ribosomal protein L3
Several transcriptomic studies have found RPL3L to be downregulated in Duchenne muscular dystrophy (DMD) [8–10], a fatal muscle wasting disorder affecting 1 in 5,000 boys [11]. DMD is caused by mutations in the DMD gene that result in the absence of the dystrophin protein, an integral component of the dystrophin-glycoprotein complex, which connects muscle fibers to the surrounding extracellular matrix [12]. Dystrophin interacts with many structural and signaling molecules at the muscle sarcolemma, hence its absence causes disruptions in multiple networks leading to a complex molecular pathophysiology that remains poorly defined and understood [13]. This has hindered efforts to develop treatments for DMD. To treat the underlying cause of DMD, dystrophin protein needs to be expressed. A promising dystrophin restoration approach is gene therapy using microdystrophin. This approach has been successful in numerous animal and preclinical studies and has now entered clinical trials in patients [14].
This article presents the first investigation into the role of RPL3L in skeletal muscles of healthy (C57) and dystrophic (mdx) mice. We investigated the developmental expression of RPL3L, we knocked down and overexpressed RPL3L in the tibialis anterior (TA) muscle via intramuscular injection of an adeno-associated virus (AAV) carrying an shRNA targeting RPL3L or RPL3L expression vector, respectively, and examined the effects on muscle function. While AAV-mediated RPL3L transgene expression did not increase protein expression likely due to unknown compensatory effects, RPL3L knockdown significantly downregulated the protein and improved muscle strength in both C57 and mdx mice. These findings show that RPL3L is important for skeletal muscle function and that its knockdown in this tissue could potentially alleviate some of the muscle force deficit observed in DMD.
Materials and Methods
All experiments were approved by the Institutional Animal Welfare and Ethical Review Body.
Generation of AAV plasmid constructs and vectors
shRNA sequences to knockdown RPL3L were designed by Benitec Biopharma and packaged into a mammalian shRNA knockdown vector driven by a U6 promoter with a GFP reporter (VectorBuilder). Knockdown was tested empirically in Human Embryonic Kidney cells (HEK293T, ATCC, Manassas, VA) cotransfected with a vector expressing RPL3L to determine the shRNA sequence that yielded maximum RPL3L knockdown (designated as shRPL3L: GAAACATCTGGAGAAAGAGAA). All experiments were approved by the Institutional Animal Welfare and Ethical Review Body.
Cell culture and AAV preparation
HEK293T cells were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 2 mM glutamine in a humidified 5% CO2 air atmosphere at 37°C. For vector preparation, a double transfection protocol was used as previously described [15]. In brief, HEK293T cells were cultured in roller bottles and transfected with the AAV-shRPL3L knockdown plasmid and the pDP9rs AAV serotype 9 helper plasmid using polyethylenimine at a ratio of 1:4. After 72 h incubation, cells were lysed and viral particles were precipitated using polyethylene glycol-2000 and purified using iodixanol step-gradient ultracentrifugation. The viral titer was determined relative to a plasmid DNA standard using real-time quantitative reverse transcription polymerase chain reaction with primers targeting the enhanced green fluorescence protein (EGFP) marker of the shRNA construct (AGCAGCACGACTTCTTCAAGTCC and TGTAGTTGTACTCCAGCTTGTGCC) and primers targeting the AAV polyA region of the shRNA construct (GAGTTTGGACAAACCACAAC and CCCCCTGAACCTGAAACATAAAATG).
In vivo experiments
Ethical and operational permission for in vivo experiments was granted by the RHUL Animal Welfare Committee and the UK Home Office. This work was conducted under statutory Home Office regulatory, ethics and licensing procedures, under the Animals (Scientific Procedures) Act 1986 (Project Licence 70/8271) C57BL/10 and mdx mice were housed with food and water ad libitum in minimal disease facilities (Royal Holloway, University of London). In brief, 8 weeks old male mice were anesthetized with isoflurane and 5 × 1010 viral particles of AAV-shRPL3L or 5 × 1011 viral particles of AAV-RPL3L-transgene were diluted in 50 μL saline and intramuscularly injected into the left TA muscle. Saline was intramuscularly injected into the right TA muscle to serve as a contralateral control. At 7 weeks postinjection, mice were anesthetized with an intraperitoneal injection of Pentobarbital/Buprenorphine solution and in situ TA muscle electrophysiology was performed. After analysis, mice were culled and TA muscles were excised from tendon to tendon, weighed, mounted in optimal cutting temperature compound and frozen in liquid nitrogen-cooled isopentane.
Muscle force measurement
Mice were put under terminal anesthesia using intraperitoneal injection of a Pentobarbital (60 mg/kg) and Buprenorphine (3 mg/kg) solution, and contractile properties of TA muscles were analyzed by in situ muscle electrophysiology using a protocol previously described [15]. In brief, clamps were used to fix the knee and foot of the mouse and the distal tendon of the TA muscle was attached to a 305B dual-mode servomotor transducer (Aurora Scientific, Aurora, Ontario, Canada) using a 4.0 braided surgical silk (Interfocus, Cambridge, UK). The sciatic nerve of the mouse was proximally cut and stimulated distally by a bipolar silver electrode using supramaximal square wave pulses of 0.02 ms duration (701A stimulator; Aurora Scientific). Isometric measurements were made at an initial length L0 (length at which maximal tension was obtained during the tetanus). Muscle length was measured using a digital calliper. Response to tetanic stimulation (pulse frequency: 10, 30, 40, 50, 80, 100, 120, 150, 180 Hz) was recorded and the maximal force was determined. A Lab-View-based DMC program (Dynamic muscle control and Data Acquisition; Aurora Scientific) was used to record and analyze data provided by the isometric transducer. Specific force (g/cm2) was calculated by dividing the maximal tetanic force by TA muscle cross-sectional area estimated using the following formula: muscle weight (g)/[TA fiber length (Lf; cm) × 1.06 (g/cm3)]. To assess the muscle resistance to eccentric contractions, a lengthening of 10% of muscle was applied for 10 consecutive contractions. Maximal (isometric) force generated after each eccentric contraction is expressed as percentage of to the initial maximal force. Afterwards, contractile measurement muscles were collected.
Western blot
Protein lysates were prepared by homogenizing muscle in radioimmunoprecipitation assay buffer (1% nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl-sulfate, 50 mM NaCl, 20 mM Tris-HCl pH 7.6, 1 mM ethylenediaminetetraacetic acid (EDTA) with complete protease inhibitor cocktail (Sigma-Aldrich) and Pierce phosphatase inhibitor) using 3 mm tungsten carbide beads with the TissueLyser system (Qiagen) for whole muscle samples, or by vortexing for muscle sections, followed by centrifugation at 16,000 g at 4°C for 10 min. The supernatant was retained and protein concentration was quantified using the DC protein assay (BIO-RAD) with a bovine serum albumin standard. The protein lysate was denatured in NuPAGE sample reducing agent and lithium dodecyl sulfate sample buffer (Invitrogen) for 10 min at 70°C before separation by gel electrophoresis. Proteins were separated on 3%–8% Tris-Acetate gels (Invitrogen) and transferred onto a 0.25 μm nitrocellulose membrane (GE Healthcare) for 1.5–2 h at 30 V constant. The membrane was blocked in 5% milk in phosphate-buffered saline (PBS) for 1 h at room temperature (RT) then stained overnight at 4°C with primary antibodies specific for vinculin raised in mouse (Sigma SAB42000080, 1/10,000 dilution), RPL3L raised in rabbit (gift from J.J. McCarthy, 1/1,000 dilution), RPL3 (Proteintech 11005-1-AP, 1/2,000 dilution), and human influenza hemagglutinin (HA) raised in rabbit (Abcam ab18181). Membranes were then incubated for 1 h at RT with appropriate secondary antibodies conjugated to LICOR IRDye 800CW [goat anti-mouse IgG (H+L) and goat anti-rabbit IgG (H+L), 1/10,000 dilution]. The LI-COR Odyssey Infrared Imaging System was used to detect signals from the membrane, with quantification using LI-COR Image Studio software.
Immunofluorescence staining and muscle histology
Frozen TA muscles were sectioned at 10 μm thickness using an OTF 5000 cryostat (Bright). Tissue sections were collected on coated glass slides (VWR) and stored at −80°C before use. For RPL3L/laminin and collagen VI/laminin immunostainings, sections were rehydrated in PBS, blocked with 10% goat serum in PBS tween 0.5% (PBST) for 1 h at RT. Afterward sections were incubated with primary antibodies. Slides were stained with primary antibodies for RPL3L (in-house polyclonal rabbit antibody generated by John McCarthy, diluted 1:200), laminin (rat monoclonal, diluted 1:800, Sigma-Aldrich L0663) and collagen VI (rabbit polyclonal, diluted 1:200, Abcam ab6588) for 1 h at RT. Secondary antibodies were Alexa-fluor (Life Biotechnologies, Paisley, UK) conjugated to 488 or 594 fluorochromes, diluted 1:200 and used for 1 h at RT. Finally, slides were stained with 1 μg/mL 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma) for 5 min at RT, washed, and mounted in Mowiol 4-88 (Sigma). For Sirius red staining, slides were air-dried, fixed in 4% paraformaldehyde, washed in water, dehydrated in 100% ethanol, and air-dried again. Slides were then stained in Sirius red solution (0.3% in picric acid) for 1 h, washed in water, fixed in 0.5% acetic acid, dehydrated in 100% ethanol, cleared in xylene and mounted in dibutyl phthalate in xylene mounting medium. Morphometric analyses were performed as previously published [16]. In brief, myofiber cross-sectional area (CSA) and the percentage of fibers with central nuclei were quantified by taking five random fields in the largest section of each muscles stained with laminin/DAPI and analyses were performed using MuscleJ software [17]. Analyses of area covered by collagen VI or Sirius red were performed by taking five random fields in sections stained by collagen VI/DAPI or Sirius red, respectively, and quantification was performed using NIH ImageJ analysis software (NIH, Bethesda, MD). All images were randomly taken from a blinded observer. Slides were imaged using a Nikon Ni-E upright microscope.
Statistical analysis
All data are presented as mean values ± error of the mean. GraphPad Prism (Version 6.07) was used for the analyses. Statistical analyses were performed using the two-tailed t-test, two-tailed paired t-test, or two-way analysis of variance (ANOVA) as appropriate. A difference was considered to be significant at *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001.
Results
RPL3L and its paralog RPL3 are developmentally regulated in skeletal muscle
To investigate the developmental expression of RPL3L and its paralog RPL3, western blot analysis was performed using TA muscles from C57 and mdx mice aged 1, 3, 7 days (1 week), 14 days (2 weeks), and finally 56 days old (8 weeks). RPL3L protein expression was up to 10.8-fold lower in 1 week old mdx compared to C57 mice (P < 0.05, Fig. 1A, B), consistent with previous studies that showed that RPL3L mRNA is significantly downregulated in dystrophin-deficient muscle [8–10]. We also observed that RPL3 protein expression was up to 4.1-fold higher in 8-week-old mdx compared to C57 mice (P < 0.05, Fig. 1A, C). This dataset displaying an inverse relationship in protein expression of RPL3L and RPL3 suggests strong postnatal developmental regulation. To assess where in the myofibers that RPL3L protein is expressed, we analyzed TA muscles of 4- and 8-week-old C57 and mdx mice through immunohistochemical staining of prepared cryosections. At these stages, myofibers in C57 muscles show a compact, organized structure with no central nucleation and a small amount of fibrotic tissue (Fig. 1D and Supplementary Fig. S1). On the contrary, in dystrophic muscles of 4-week-old mice, several clusters of small centrally nucleated fibers (CNF) are observed together with large areas of fibrotic tissue (Fig. 1D and Supplementary Fig. S1). In TA muscles of 8-week-old mdx mice, a significant reduction in regeneration and fibrosis was detected relative to C57 TA muscle (Fig. 1D and Supplementary Fig. S1). RPL3L protein was detected at both time points in all muscles analyzed mainly in the subsarcolemmal region or at the level of sarcolemma (Fig. 1D). In TA muscles of 4 weeks old mdx mice, only the large myofibers stained positive, while clusters of small regenerating myofibers did not express RPL3L (Fig. 1D) for RPL3L, suggesting that the reduction in protein expression displayed by mdx muscles may be due, at least partially, to the lack of expression in the smaller regenerating fibers. In TA muscles of 8 weeks old C57 and mdx mice, all fibers express the protein although lower expression was detected in mdx muscles.
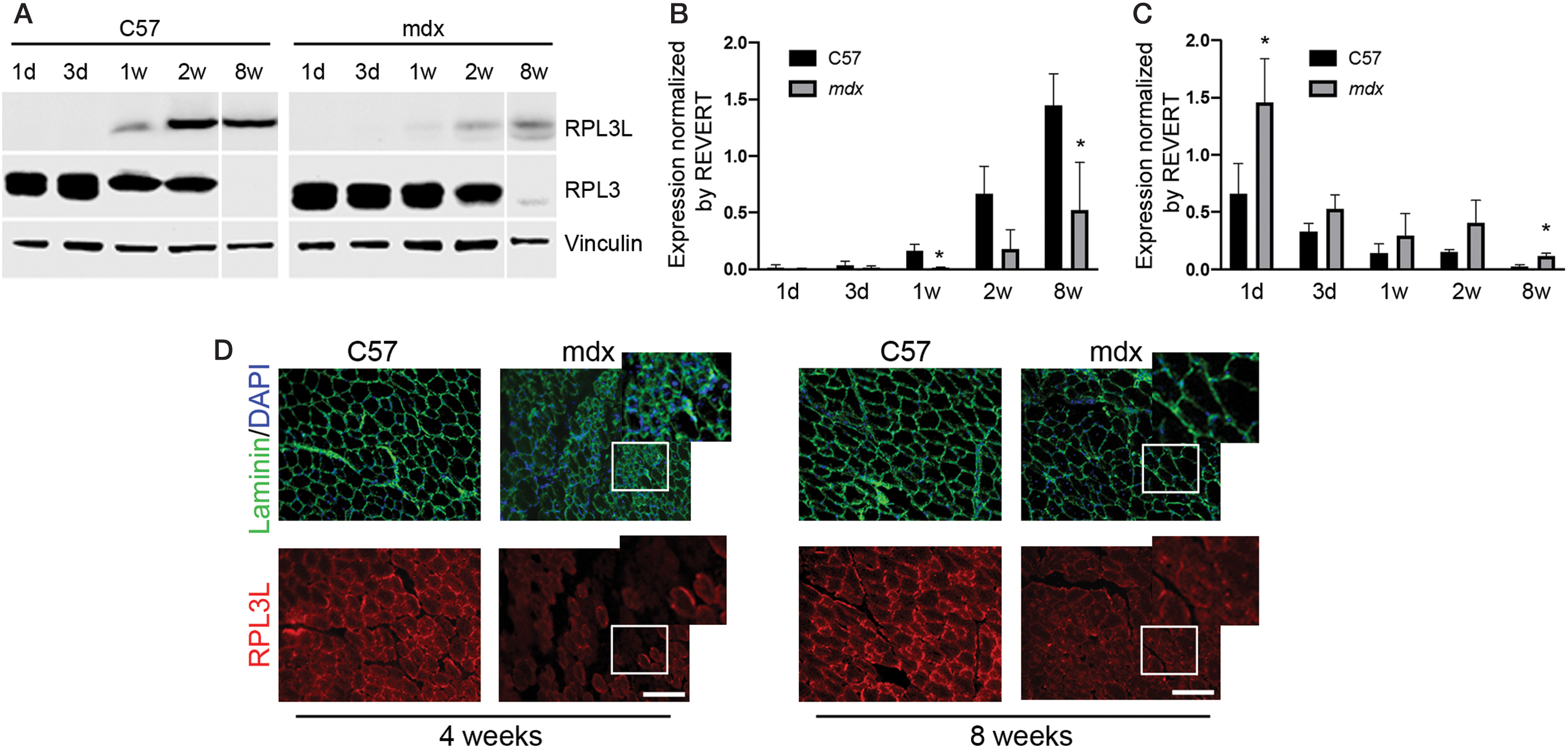
Developmental expression of RPL3L and RPL3.
RPL3L overexpression does not affect muscle function while its knockdown significantly improves muscle function in C57 and mdx mice
To determine the function of RPL3L in skeletal muscle, we tried to either overexpress or knockdown RPL3L in the TA muscle of C57 and mdx mice. Specifically 5 × 1011 viral particles of AAV-9 encoding a transgene to overexpress RPL3L (AAV-RPL3L) or 5 × 1010 viral particles AAV9 encoding a shRNA to knockdown RPL3L (AAV-shRPL3L) were injected into the left TA, with the contralateral muscle injected with an equal volume of saline in eight C57 and six mdx mice at 6 weeks of age. Seven weeks after injection, TA muscle function was assessed by in vivo muscle electrophysiology and the muscles were harvested. Expression of RPL3L construct in C57 and mdx was confirmed by western blot analyses as suggested by HA tag expression (Supplementary Fig. S2A). However this did not result in RPL3L protein overexpression (Supplementary Fig. S2A, B), neither in significant changes in maximal tetanic or maximal specific force nor in resistance to eccentric contractions (Supplementary Fig. S2C–E). Accordingly, muscle length, muscle mass, and muscle CSA were also not significantly affected (Supplementary Fig. S2F–M). On the contrary, muscle electrophysiological analysis revealed a significant improvement of muscle strength and resistance to fatigue in C57 and mdx mice following RPL3L knockdown. RPL3L knockdown in C57 mice showed a significant 1.20-fold increase in specific force (P < 0.05, Fig. 2A), while in mdx mice AAV-shRNA treatment resulted in a significant 1.45-fold increase (P < 0.05) (Fig. 2B). RPL3L knockdown conferred a protective effect against eccentric contraction induced damage (normally seen in dystrophic muscle [18,19]) with treated muscles showing greater resistance compared to control dystrophic muscles (keeping at least 1.7-folds the resistance of control muscles from the fifth to the last contraction, P < 0.001 to P < 0.05, Fig. 2C).
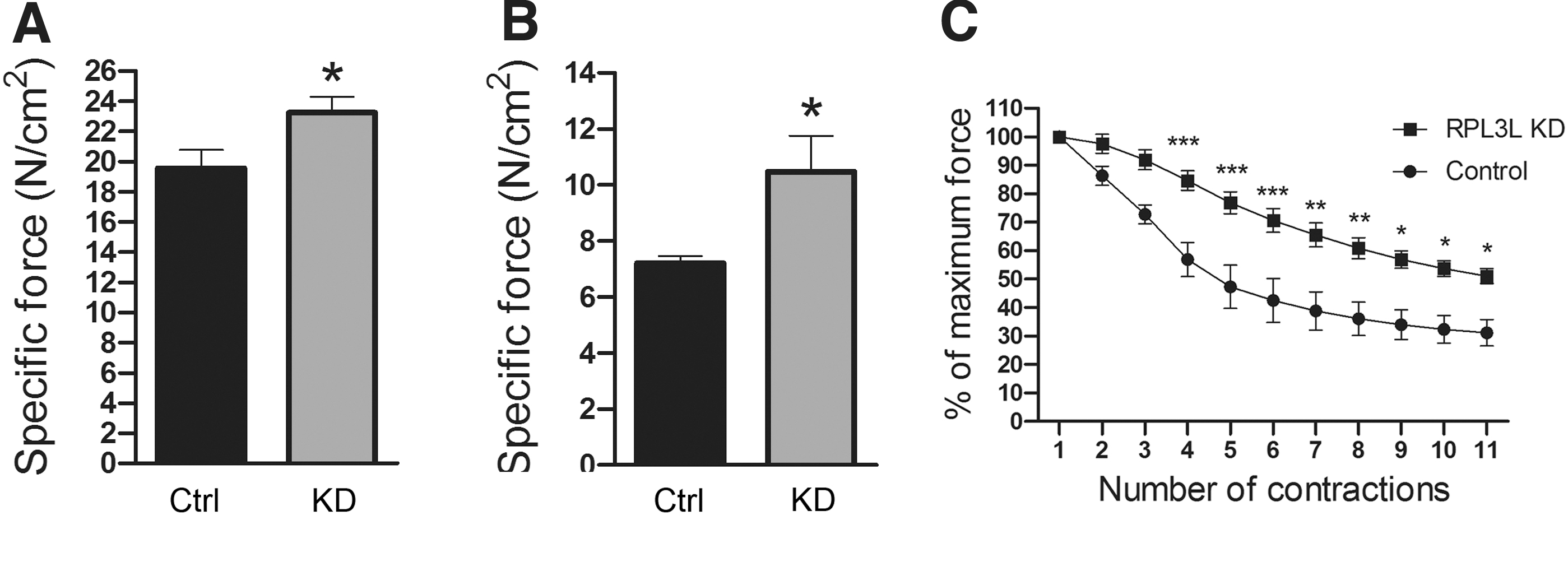
In situ TA muscle electrophysiology following RPL3L knockdown in C57 and mdx.
Analysis of TA muscles showed successful knockdown of RPL3L in the C57 and mdx mice compared to controls (Fig. 3A–D) with significant reduction in RPL3L protein expression to 21.70% ± 0.04% of control levels in C57 mice (P < 0.01, Fig. 3A, B) and 27.90% ± 0.02% of control levels in mdx mice (P < 0.05, Fig. 3C, D). Significant protein inhibition was also observed by immunofluorescence in both C57 and mdx muscles (Fig. 3E, F).
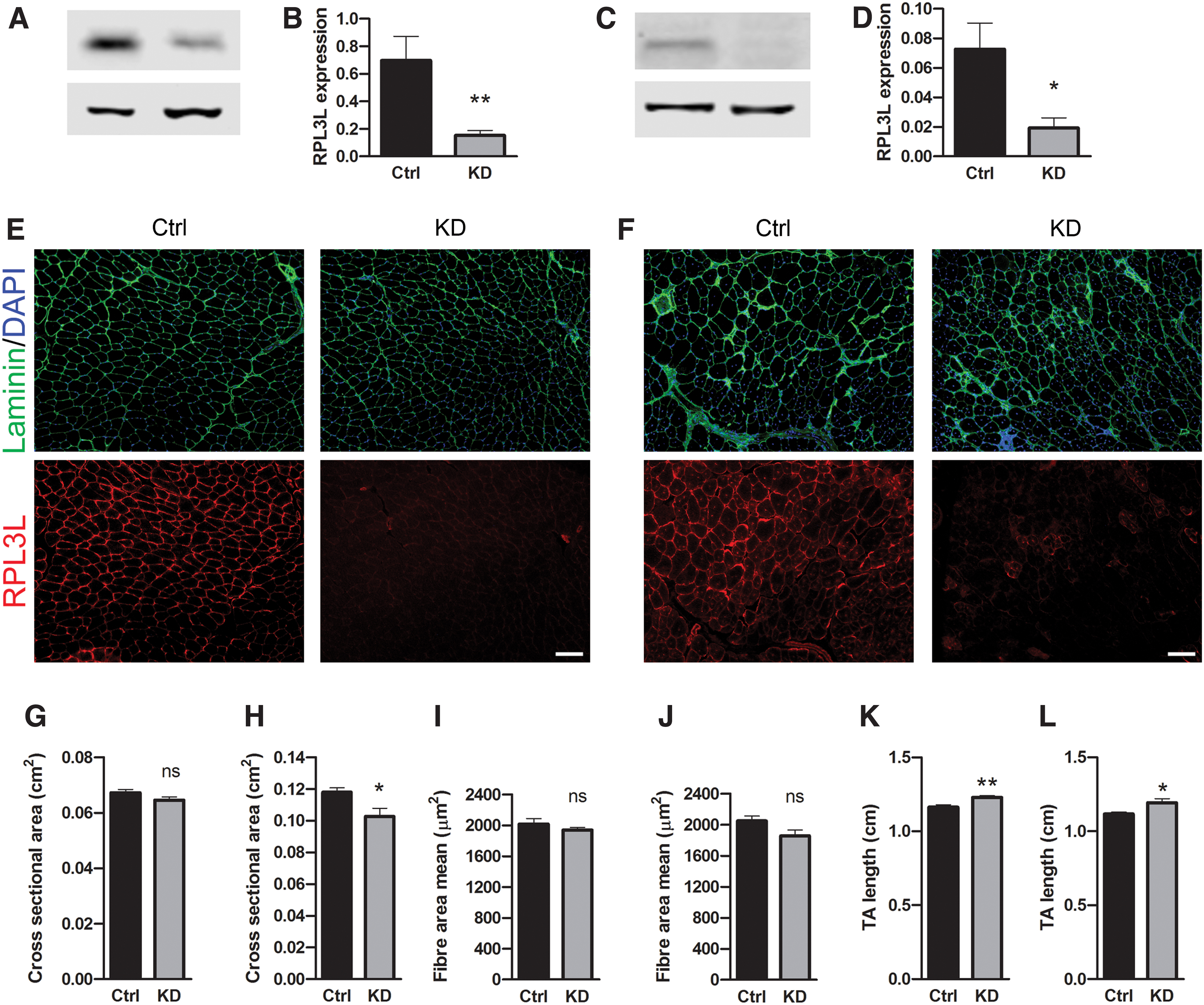
RPL3L knockdown in C57 and mdx.
No significant differences were observed in TA mass of either C57 (P = 0.60) or mdx (P = 0.22) muscles between control and knockdown groups (data not shown). No difference was observed for the TA CSA of whole C57 muscles treated with shRNA or saline (Fig. 3G), but TA cross-sectional area significantly decreased in mdx mice (P < 0.05) (Fig. 3H). Myofiber CSA reduction in both C57 and mdx mice was not significant (Fig. 3I, L), although the difference in mdx mice was larger and closer to achieving significant difference (11.80% ± 0.04% decrease in mean fiber area, P = 0.07), (Fig. 3L). Notably, TA length was significantly longer following RPL3L knockdown in both C57 (P < 0.01, Fig. 3M) and in mdx mice (P < 0.05, Fig. 3N). Since RPL3L knockdown significantly improved muscle force in mdx mice, we assessed the percentage of CNF and the fibrotic tissue deposition. However, neither the percentage of CNF (Supplementary Fig. S3A, B) nor the area covered by collagen VI or stained by Sirius red (Supplementary Fig. S3A, C–E) significantly changed in muscles where RPL3L is knocked down compared to saline-treated controls. These data suggest that while RPL3L overexpression does not affect muscle function and morphology, its inhibition significantly changes muscle shape and increases skeletal muscle strength. The improvement in muscle force is not due to changes in myofiber nucleation or fibrotic tissue deposition.
Discussion
Our data confirmed that RPL3L and its paralog RPL3 are developmentally regulated and display an inverse relationship. Low expression of RPL3L and high expression of RPL3 coincide with rapid muscle growth during the neonatal period, while high expression of RPL3L and low expression of RPL3 occur once the rate of growth slows down in adulthood. Accordingly, we found that the clusters of small regenerating fibers usually detectable in large numbers in muscles of dystrophic mice at very young age do not express RPL3L. Once the tissue degeneration-regeneration processes slow down and myofibers achieve a more mature stage, RPL3L is expressed by all myofibers. These observations are consistent with previous work by Chaillou et al. who reported that RPL3L is downregulated and RPL3 is upregulated in processes leading to hypertrophy. In response to a hypertrophic stimulus, higher number of myogenic cells, where RPL3L is downregulated, fuse with the preexisting fibers to increase the muscle size. The authors showed that induction of RPL3L expression impairs myotube growth and myoblast fusion and proposed that RPL3L is a negative regulator of muscle growth [7]. However, we did not observe a significant increase in TA mass or CSA following RPL3L knockdown. On the contrary, a significant increase in myofiber length was observed. These data suggest that even though RPL3L is downregulated in hypertrophy [7], this is not caused by RPL3L knockdown in vivo at least when the protein inhibition is performed postnatally.
Given that RPL3L is expressed during periods of slow growth and the hypothesis that RPL3L is a negative regulator of muscle growth [7], it is possible that RPL3L is responsible for slower translation of muscle-specific mRNA transcripts, compared to RPL3. Following from this hypothesis, downregulation of RPL3L would result in upregulation of muscle-specific proteins. Techniques such as polysome and ribosome profiling are required to confirm this. These experiments were not feasible in this study due to the limited quantity of AAV-injected muscle, but this could be overcome by generation of an RPL3L-knockout mouse.
The most striking result of this study was that RPL3L knockdown enhanced muscle function in both C57 and mdx animals. This is somehow expected if RPL3L is a true negative regulator of muscle growth, as already observed for example for myostatin [20]. It has been shown in multiple studies that while myostatin, a member of the TGFβ family and negative regulator of muscle growth, is indeed downregulated in dystrophic muscles [21], its further inhibition improves muscle pathology, in particular once a parallel treatment to restore dystrophin is provided [16,22–24]. This is further confirmed by the observation that RPL3L overexpression in dystrophic muscles resulted in neither significant improvement nor impairment of muscle function that also leads to the conclusion that the downregulation of RPL3L in DMD does not contribute to the DMD pathophysiology.
It is possible that downregulation of RPL3L is a protective mechanism triggered to try and improve muscle function in dystrophic conditions that, however, is not sufficient to counteract the many layered effects and complex pathophysiology of dystrophin-deficiency. Therefore, a further knockdown of RPL3L by AAV-shRPL3L injection is beneficial to improve muscle function in mdx. These results suggest that RPL3L inhibition could be used as a therapeutic strategy for treatment of skeletal muscles in DMD. Related to this, it would be interesting to analyze the RPL3L expression in dystrophic cardiac muscle as well because a correct expression of RPL3L is important to maintain the functionality of the cardiac muscle [6]. Furthermore, it would be interesting to examine the levels of RPL3L in other muscular dystrophies to verify that a similar RPL3L endogenous downregulation exists.
As expected, due to the short time point used in our experiments, RPL3L knockdown did not result in change in fibrosis deposition or myofiber central nucleation. However, we cannot exclude that, in the long term, RPL3L reduction could affect muscle regeneration processes. RPL3L knockdown resulted in a significant increase in TA length in both C57 and mdx. Although there are no reported links between increased muscle length and increased specific force, there is, however, a link between decreased sarcomere length and decreased specific force [25]. We speculate that RPL3L knockdown can affect sarcomeres in such a way as to improve muscle function possibly affecting the muscle-specific translation of proteins determinant of sarcomere length, such as titin [25]. This is indeed a long protein that may need a quick translation, like the one provided by RPL3 that is supposed to be upregulated when RPL3L is inhibited. It remains unclear how resistance to eccentric contraction-induced damage in mdx is improved. In summary, we show that RPL3L knockdown can enhance muscle function in healthy muscles and propose that endogenous RPL3L downregulation seen in DMD is a protective mechanism and attempt to enhance muscle function and counteract the dystrophic phenotype.
Footnotes
Acknowledgments
The authors thank Benitec Biopharma for designing shRNAs targeting RPL3L, Ornella Cappellari for assistance with in vivo muscle electrophysiology, and Emma Popescu and Nicola Sanderson for care and maintenance of animals.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
B.R.K. was funded by the Neurological Foundation of New Zealand.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.