
Abstract
The ABCA4 gene, involved in Stargardt disease, has a high percentage of splice-altering pathogenic variants, some of which cause complex RNA defects. Although antisense oligonucleotides (AONs) have shown promising results in splicing modulation, they have not yet been used to target complex splicing defects. Here, we performed AON-based rescue studies on ABCA4 complex splicing defects. Intron 13 variants c.1938-724A>G, c.1938-621G>A, c.1938-619A>G, and c.1938-514A>G all lead to the inclusion of different pseudo-exons (PEs) with and without an upstream PE (PE1). Intron 44 variant c.6148-84A>T results in multiple PE inclusions and/or exon skipping events. Five novel AONs were designed to target these defects. AON efficacy was assessed by in vitro splice assays using midigenes containing the variants of interest. All screened complex splicing defects were effectively rescued by the AONs. Although varying levels of efficacy were observed between AONs targeting the same PEs, for all variants at least one AON restored splicing to levels comparable or better than wildtype. In conclusion, AONs are a promising approach to target complex splicing defects in ABCA4.
Introduction
Stargardt disease (STGD1) is an autosomal recessive macular disease characterized by progressive degeneration of central vision accompanied by accumulation of autofluorescent lipofuscin flecks and distinctive peripapillary sparing.1,2 The genetic causes of disease are biallelic variants in the ABCA4 gene, encoding the ATP binding cassette subfamily A member 4 protein. 3 ABCA4 is a transmembrane protein that is mainly located in the lamellae of cones and outer segment discs in rods,4,5 although expression in the retinal pigmented epithelium (RPE) has also been reported. 6 It has a crucial role in eliminating toxic by-products of the visual cycle, specifically by importing N-11-cis-retinylidene-phosphatidylethanolamine to the lumen side of outer segment discs. 7 Consequently, ABCA4 dysfunction causes the accumulation of phosphatidylethanolamine and other toxic derivates, such as phosphatidyl-pyridinium bisretinoid (A2PE) and its photoisomer A2E in the outer segments of photoreceptors. Upon phagocytosis of the outer segments, these compounds become part of lipofuscin deposits in the RPE causing progressive atrophy of the central retina that manifests with loss of vision.7,8
Around 1,247 of the >2,200 known unique ABCA4 variants (http://www.lovd.nl/ABCA4) have been classified as pathogenic or likely pathogenic after assessment by adapted ACMG/AMP guidelines. 9 ABCA4 variants can be also subdivided based on their severity using a phenotype–genotype model. 10 This second method of classification is key to elucidate the correlation between different variant combinations and the spectrum of phenotypic manifestations observed in patients with ABCA4-associated retinopathy. The main severity categories are benign, mild, moderately severe, and severe/null, which reflect the amount of residual function of the mutant protein. Different combinations of variants correspond to different disease severities from early-onset STGD1 or panretinal cone-rod dystrophy (severe|severe) to classical STGD1 (moderately severe|moderately severe or severe|mild), to late-onset STGD1 associated with the combination of a severe and a mild, low penetrant variant. Partial loss of allele functionality caused by a moderately severe or mild allele can thus lead to disease manifestation, as evidenced in the relatively large number of non-protein truncating pathogenic variants described in ABCA4 (61% of all ABCA4 variants). 11 Splicing alterations have been extensively reported as a relevant pathogenic mechanism involved in ABCA4-associated retinopathies.12–16 Around 10% of known ABCA4 variants are deep intronic (DI) or noncanonical splice site variants, whose effect encompass complete or partial exon skipping, complete or partial intron retention or pseudo-exon (PE) inclusion. Exonic single nucleotide variants (SNVs) outside the splice sites can also result in splice defects. 17 Similar to missense variants, splicing alterations can lead to different amounts of residual activity of the ABCA4 protein depending on the percentages of wild-type and mutated transcript produced. The comparatively high frequency of splice-altering variants in ABCA4 renders them interesting targets for therapeutic intervention, even more so considering that even partial rescue could be beneficial. One promising strategy for targeting aberrant splicing is the use of antisense oligonucleotides (AONs), which have been shown to efficiently restore correct splicing in ABCA412,15,18–20 and other genes implicated in inherited retinal diseases, such as CEP290,21–25 USH2A, 26 CHM, 27 and OPA1. 28 AONs are single-stranded RNA molecules designed to target specific splicing elements, e.g., cryptic splice sites, enhancer/silencer sequences, to promote correct splicing at the pre-mRNA level by blocking recognition of spliceosome binding sites. Despite the current popularity of AON therapeutic approaches, their effectiveness on complex splicing defects, where multiple alterations are present in the transcript, remains to be assessed. In ABCA4, such complex defects have previously been described in different intronic regions, i.e., intron 1313,29 and intron 44. 13
In this study, we assessed the effectiveness of AON rescue for selected ABCA4 complex splicing defects. In particular, four variants in intron 13 leading to compound PE inclusions and one variant in intron 48 leading to multiple PE inclusions and exon skipping were selected to test six AONs designed to target the defects.
Materials and Methods
Variant selection
Variants for the AON rescue study were selected considering previously characterized variants leading to unusually complex splicing defects. Intron 13 was of particular interest as multiple variants leading to the same or similar splicing defect, characterized by the inclusion of two nonadjacent PEs in a portion of the mutant transcript, are known. Apart from three previously described SNVs, i.e., c.1938-514A>G, c.1938-621G>A, 13 and c.1938-619A>G 29 (Fig. 1B and C), a fourth novel variant was later included in the study (c.1938-724A>G, details in Supplementary Data S1and Supplementary Table S1) as it was situated in the same hotspot and confirmed to lead to a similar splicing defect (Fig. 1A). Finally, c.6148-84A>T, which is situated in intron 44, was selected as previous studies showed multiple splicing defects, including concurrent PE inclusion and skipping of adjacent exons (Fig. 1D).
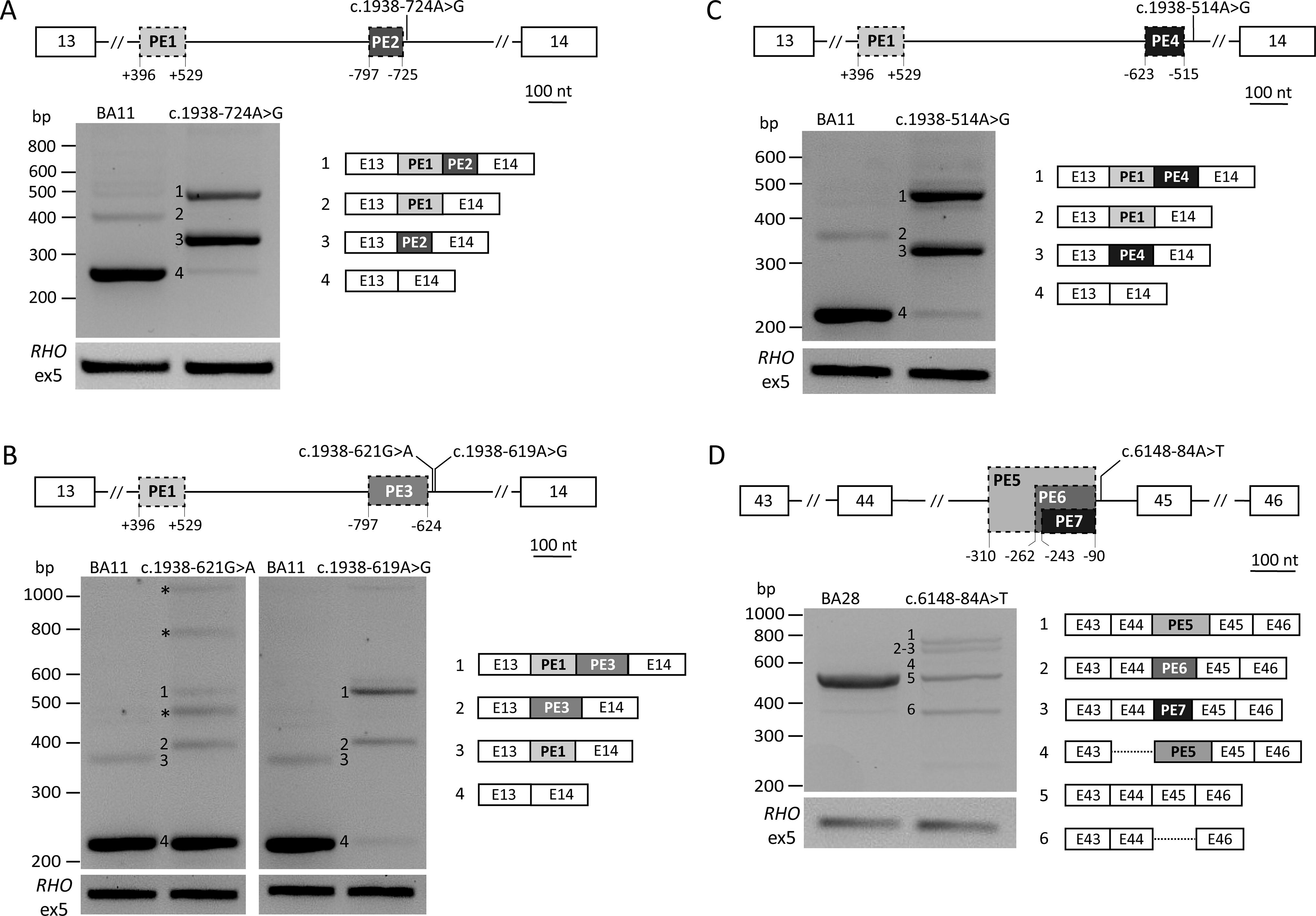
Intron 13 and 44 variants resulting in complex splice-altering defects. Each panel depicts the effects of specific variants. On top, a schematic of the affected gene region depicts the relative position of the multiple defects. White boxes represent exons, while gray boxes with stapled lines represent PEs. Numerals depict the intronic positions of the start and end of PEs. RT-PCR amplification was performed using primers situated in the first and last exon shown, and amplification of RHO exon 5 was used as transfection control. In the gel images, bands for which the exact sequence is known are numbered and the corresponding defect is illustrated on the right, dotted lines denote exon skipping. Asterisks in gel images denote PCR artifacts.
AON design
A total of five novel AONs were designed for the regions of interest (Table 1), according to previous guidelines.30,31 All four intron 13 variants (c.1938-724A>G, c.1938-621G>A, c.1938-619A>G, and c.1938-514A>G) lead to the inclusion of an upstream PE (PE1), which was targeted with a previously described AON 15 with adjustments in the sugar moiety (from 2′-O-methyl to 2’-O-methoxyethyl), whereas the AON sequence was not modified. Three AONs were used to target the region of interest in intron 13, as the location of PE2 (c.1938-724A>G), PE3 (c.1938-621G>A, c.1938-619A>G), and PE4 (c.1938-514A>G) is either overlapping (PE2-PE3) or contiguous (PE3-PE4). These AONs were designed to target the cryptic splice sites of the PEs directly (AON-B and AON-C) or the adjacent region (AON-D) (Fig. 2A). AON-C binds to the region that harbors the variants c.1938-621GA and c.1938-619A>G. The oligonucleotide was designed to be specific for c.1938-621G>A, as avoiding mismatches was not possible. The complex splicing defects caused by variant c.6148-84A>T were targeted by two AONs, AON-E targeted the cryptic splice acceptor site (SAS) of the smaller PE6 and AON-F the cryptic splice donor site (SDS) shared by both PE5 and PE6 (Fig. 3A). All AONs had a phosphorothioate (PS) backbone, not controlled for stereochemistry, and a 2’-O-methoxyethyl sugar modification. As negative control, a scrambled oligonucleotide was used. The oligonucleotides were purchased from Eurogentec (Liege, Belgium), resuspended at 100 µM in phosphate-buffered saline (PBS), and used at a final concentration of 0.5 µM.
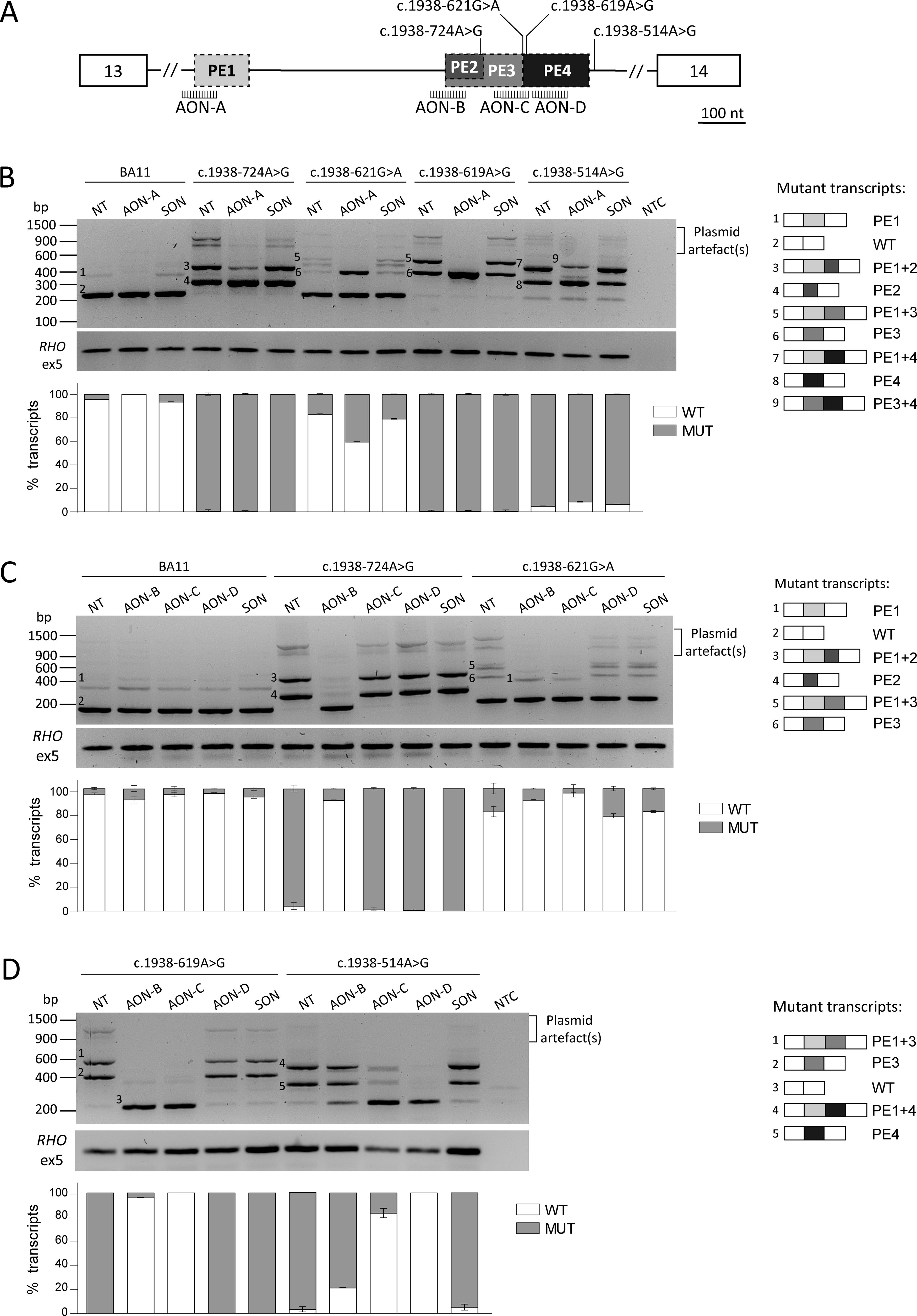
AON rescue of intron 13 splicing defects.
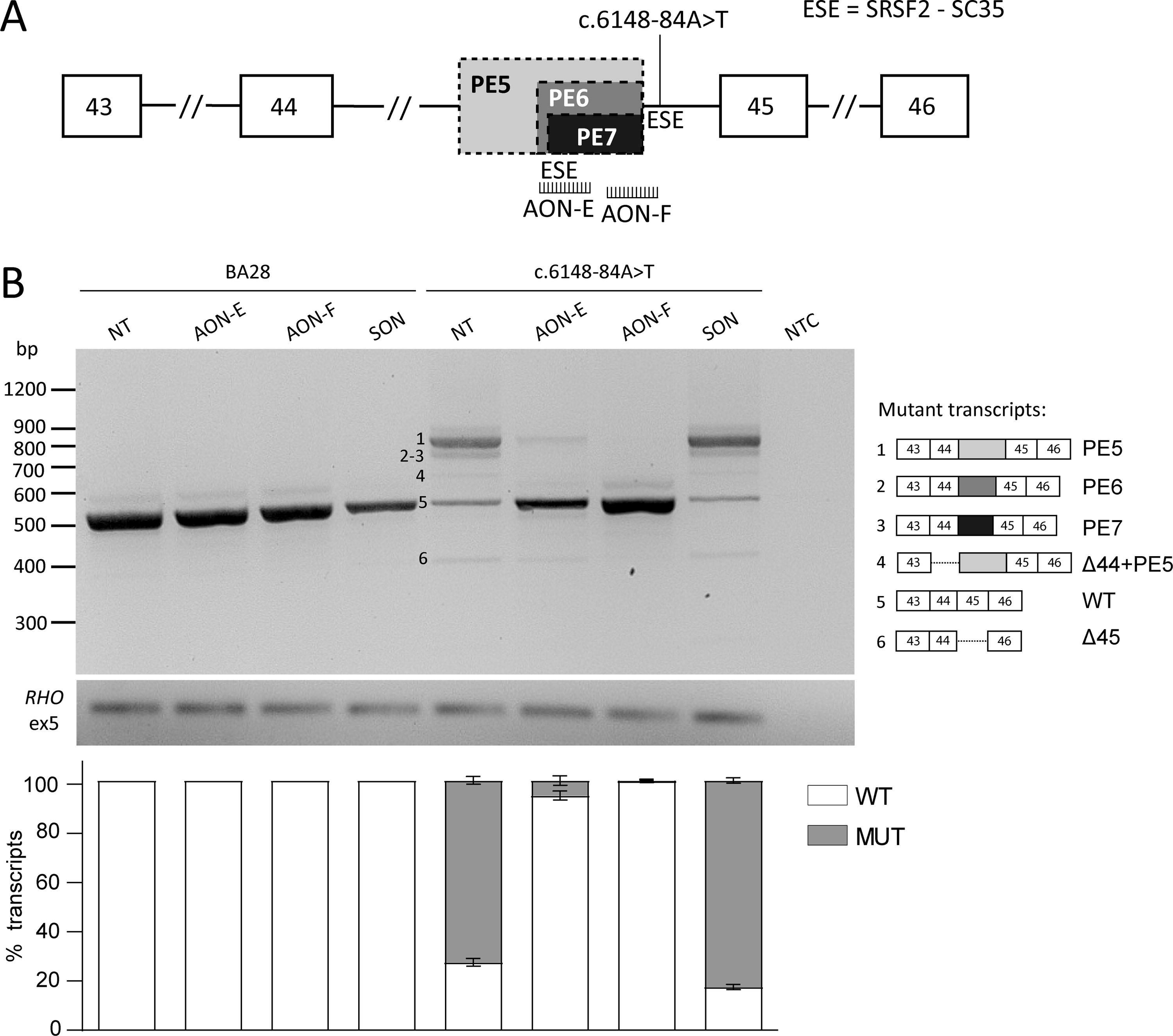
AON rescue of intron 44 splicing defects.
Sequence and Characteristics of the Antisense Oligonucleotides Used to Target Complex Splicing Defects
AON, antisense oligonucleotide; NT, nucleotides; PE, pseudoexon; SON, scrambled oligonucleotide; Tm, melting temperature.
Midigene-based splice assay and AON efficacy
The efficacy of the AONs was assessed in vitro by splice assays of two midigene constructs (BA11 and BA28, Supplementary Fig. S1). Briefly, wild-type (WT) and mutant midigenes were transfected in HEK293T cells and subsequently treated with 0.5 µM of AON, scrambled oligonucleotide (SON) or were left untreated as controls. After reverse transcription polymerase chain reaction (RT-PCR), transcripts were analyzed by agarose electrophoresis gel and Sanger sequencing (Supplementary Figs. S2–S6), followed by Fiji software densitometry analysis (Supplementary Table S1 and S2). 32 All primers used can be found in Supplementary Table S3. The complete description of the splice assay protocol performed is reported in Supplementary Data S1.
Results
Deep-intronic variant c.1938-724A>G results in a complex splicing defect
The novel variant c.1938-724A>G was identified in a Canadian proband who was selected for complete ABCA4 locus sequencing as only one pathogenic variant, c.71G>A;p.(Arg24His), was found in the coding regions of ABCA4. SpliceAI scores for c.1938-724A>G showed a strong SDS gain (DG = 0.58, at position c.1938-724) and a strong SAS gain (AG = 0.61, at position c.1938-797), suggesting a high likelihood that the variant could lead to a PE inclusion (Supplementary Table S4A). Consequently, splice assay analysis of this variant revealed multiple splicing alterations (Fig. 1A). The most abundant transcript, corresponding to 72% of the total RT-PCR product, carried a short PE of 73 nt (PE2), as predicted by SpliceAI. The second main transcript (24% of total RT-PCR product) corresponded to the inclusion of PE2 in combination with the known upstream PE1, which was previously described both on its own and in combination with different PEs.13,15,29 Both defects introduce a premature stop codon and are expected to trigger nonsense-mediated decay. As aberrant splicing was observed in 96% of the total transcript, the variant was classified as severe, following the genotype-phenotype correlation model for ABCA4-associated retinopathies. 11 The updated RNA and protein notations (hg19, NM_000350.3) are r.[1937_1938ins1938-797_1938-725,[1937_1938ins1937+396_1937+529;1938-797_1938-725]] and p.[Phe647Alafs*24,Phe647Serfs*24], based on semi-quantification of the cDNA products.
The newly identified variant c.1938-724A>G was included in the AON study design of intron 13 variants because its defects showed significant similarities with the variants that had already been selected for the study. First, PE2 partially overlaps with the longer PE3 (174 nt) that occurs in the mutant transcripts resulting from variants c.1938-621G>A and c.1938-619A>G. In particular, PE2 inclusion is triggered by the creation of a novel SDS at position c.1938-725 together with the activation of the cryptic SAS at position c.1938-797, which corresponds to the SAS of PE3. Second, the inclusion of PE1 was observed in all other intron 13 variants for which an AON approach was designed.
ABCA4 intron 13 complex splicing defect rescue
The experimental design for rescuing the selected intron 13 variants was developed considering the complex nature of the observed splicing defects. The two regions affected by splicing abnormalities were targeted separately because of the distance between the upstream PE1 and the cluster of other PEs, with the possibility of combining different AONs to restore correct splicing, if needed. Figure 2A shows a graphical representation of the three different PEs observed and how they are positioned relative to each other.
The inclusion of PE1 was targeted using AON-A (Table 1), which was previously proven to efficiently prevent PE1 insertion in midigene constructs carrying the variant c.1937+435C>G. 15 Here, we tested the effect of AON-A on complex splicing defects, where PE1 was present only in a portion of the aberrant transcript. In three of the tested variants, i.e., c.1938-621G>A, c.1938-619A>G and c.1938-514A>G, AON-A completely blocked PE1 inclusion, while for c.1938-724A>G only a partial effect was observed, as the PE1+PE2 (fragment 2) insertion was still clearly visible (Fig. 2B). Nevertheless, in all cases, there was no rescue effect observed after delivering AON-A as the levels of WT were not strongly impacted, despite the abolition of PE1 inclusion. For c.1938-514A>G, the levels of correct transcript slightly increased after treatment (from 5% to 8.6%). Nevertheless, the level of WT product remained too low to consider this as a potential beneficial effect. At the same time, a new band (fragment 8) was consistently observed in the replicates of c.1938-514A>G treated with AON-A that, upon sequencing, was revealed to correspond to the combined inclusion of PE3 and PE4, which was not observed before (Fig. 2B and Supplementary Fig. S7). On the contrary, in the case of the mild variant c.1938-621G>A, a considerable decrease of the correct transcript was observed (from 82.7% to 59.4%), where all the new aberrant product corresponded to PE3 inclusion alone. These results are likely explained by the complex landscape around the variants themselves and not because of the AON design, as AON-A efficiently excluded the PE in the sample transfected with the WT construct (BA11), where a slight amount of PE1 was also observed.
The region where PE2 to PE4 are clustered was targeted using three AONs (AON-B, AON-C, and AON-D) (Fig. 2A). AON-B targeted the SAS shared by PE2 and PE3 and proved to efficiently rescue three of the tested variants, i.e., c.1938-724A>G, c.1938-621G>A, and c.1938-619A>G. In particular, AON-B was the only AON that resulted in 86.2% exclusion of the short PE2, reaching a total of 90.4% correct transcript. This can be explained by the fact that it is the only AON directly binding PE2 and interfering with the recognition of the PE. Inclusion of PE3 was also efficiently blocked by AON-B, reaching 90.1% and 96.4% of correct transcript for c.1938-621G>A and c.1938-619, respectively (Fig. 2B and C). In both cases, the remaining aberrant splicing consisted of PE1 inclusion. Interestingly, the amount of PE1 observed after AON-B treatment was comparable with the levels in the WT sample treated with the same AON (c.1938-621G>A) or even lower (c.1938-619A>G). Similar trends of PE1 exclusion were observed for the other AONs tested in this study. Finally, PE4 and its corresponding variant c.1938-514A>G are located downstream of the target region of AON-B, thus no strong impact was expected after treatment. But, surprisingly, we observed a substantial reduction of aberrant transcript with 18.2% splicing correction (Fig. 2D). AON-C binds to the region containing the dual cryptic splice site with the canonical nucleotides AG/GT at positions c.1938-625_1938-622. This splice site is strengthened and recognized as an SDS in the presence of variants c.1938-621G>A and c.1938-619A>G (PE3), while it acts as the SAS of PE4 when in combination with the creation of a novel cryptic splice site by the variant c.1938-514A>G. Considering this, AON-C was designed to overlap the junction sequence between PE3 and PE4 with the aim of targeting both PEs. Unfortunately, it was not possible to design a variant-independent AON-based approach for this region as c.1938-621G>A and c.1938-619A>G are directly adjacent to the splice site. AON-C was designed to bind from c.1938-639 to 1938-620, while this allowed to avoid c.1938-619A>G, the AON was specific for c.1938-621G>A. Treatment with AON-C for c.1938-619A>G and c.1938-621G>A efficiently targeted both PE3 inclusion alone and the inclusion of both PE1 and PE3 (Fig. 2C and D). Interestingly, c.1938-619A>G was completely rescued by the AON, despite the mismatch present in the sequence. The rescue effect of AON-C on PE4 (caused by variant c.1938-514A>G) was lower, but still reached 80.1% correction. Finally, AON-D was designed to target PE4 without directly targeting its splice sites to avoid the regions containing the previously described variants. The AON was ineffective for rescue of PE2 and PE3, but was the only AON that reached complete abolition of the splicing defect caused by c.1938-514A>G.
Overall, for each variant, there was at least one AON that successfully corrected the splicing defect: AON-B for variant c.1938-724A>G, AON-B and AON-C for c.1938-621G>A and c.1938-619A>G, and AON-D for c.1938-514A>G. Most notably, in all these instances, the use of a single AON was enough to eliminate both the PE adjacent to the variant but also to block inclusion of the upstream PE1 to levels comparable to the WT control, or even lower. No changes in splicing were observed when the SON was delivered, indicating that the effect was directly promoted by the AON.
ABCA4 intron 44 complex splicing defect rescue
The intron 44 variant c.6148-84A>T was first described by Khan and colleagues, 13 and characterization of the splicing defect by midigene assays revealed a complex array of aberrant splicing that included the skipping of exon 45, inclusion of a 173-nt PE (PE6 in Fig. 1D), the inclusion of a longer PE of 221 nt (PE5 in Fig. 1D) on its own and concurrently to the skipping of exon 44. During this study, an additional PE (PE7 in Fig. 1D and Supplementary Fig. S2) of 154 nt was detected. All PEs share the same cryptic SDS at position c.6148-90, which was strengthened in the presence of the downstream variant, but made use of different cryptic SASs that are naturally present in intron 44. The two AONs selected to target these complex splicing defects were designed to bind to the common SDS of the PEs (AON-F) or the region containing the SAS of the shorter PEs (AON-E), which additionally overlaps with a strong exonic splicing enhancer (ESE) motif (Fig. 3A). Both AONs achieved high correction levels, i.e., 93.9% and 99.7%, after treatment with AON-E or AON-F, respectively. The aberrant splicing observed in AON-E-treated cells corresponded mainly with PE5 inclusion and in a smaller percentage to exon 45 skipping, while neither were observed after treatment with AON-F (Fig. 3B).
Discussion
Splice-altering variants in ABCA4 have become an attractive target for the development of novel therapies, particularly in the field of RNA splicing modulation. The effectiveness of AON molecules in restoring correct splicing in ABCA4 has been extensively demonstrated12,15,18–20 but remained unclear in complex splicing defects. In this study, we assessed the rescue efficacy of AONs in rescuing complex defects in two intronic regions of ABCA4, i.e., intron 13 and intron 44.
Three known and one novel intron 13 variants were selected as they caused complex splicing defects characterized by the inclusion of an upstream PE in a portion of the aberrant transcripts. Interestingly, this upstream inclusion cannot be explained solely by the presence of a pathogenic variant, as these are located more than 500 nt downstream. In addition, PE1 inclusion was consistently observed in the WT construct, congruently with its naturally occurring inclusion observed in photoreceptor precursor cells (PPCs) derived from a healthy individual 13 and in RNA extracted from human donor eyes. 33 Taken together, these observations make a compelling argument for the reclassification of this insertion as a poison exon (PoE) rather than a PE, where a PoE is defined as a highly conserved alternative exon that contains an in-frame stop codon leading to nonsense-mediated decay independently of any pathogenic variation.34,35 In addition, the 3’ end of the PoE presents a dual splice site (AG/GC), which has been previously suggested to act as a ratchet point 36 for recursive splicing. 13 We hypothesize that the level of PoE inclusion could be dependent on alternative and recursive splicing events (Supplementary Fig. S8A) and alterations in the splicing signaling landscape of intron 13 impact its regulation. The four intron 13 SNVs in this study are clustered in a downstream region containing several cryptic splice sites, where their strengthening is likely to impact the recognition of the upstream ratchet point, causing the observed increase in PoE inclusion in aberrant transcript compared with the WT low levels (Supplementary Fig. S8B). On the other hand, the AONs targeting the cryptic splice sites in the downstream PEs are not only directly targeting the pathogenic variants but might be indirectly strengthening the recognition of the upstream dual-splice point, by decreasing the competition between available splice sites in the downstream region. This could lead to the decreased or even null levels of PoE inclusion observed after treatment with AONs (Supplementary Fig. S8C). At the same time, if inclusion of this PoE is a naturally occurring and regulated event, this could explain why treatment with AON-A was observed to lead to counterproductive additional missplicing in some of the variants studied, such as c.1938-514A>G. Careful examination of the alterations between WT, mutant, and AON-treated splicing landscapes leads us to advance the hypothesis that recursive splicing mechanisms are involved in the regulation of intron 13 splicing and the putative ratchet point at the SDS of the PoE has a crucial role in this mechanism. In recent years, it has been suggested that alternative splicing events have a role in transcript regulation and quality control. 37 In particular, a recent study 38 reports the presence of ratchet points at the distal end of cassette exons, leading to exon exclusion. The dual splice site at the SDS of PoE might be acting in an analogous way. Interestingly, other instances of ratchet points localized at the splice site of putative PoE have been reported outside of ABCA4. 39 While our AON rescue studies indirectly support the hypothesis that recursive splicing is implicated in the regulation of PoE1 and intron 13 splicing, follow-up studies, e.g., nascent RNA level analysis, are needed to further substantiate the proposed mechanism.
Independently of these observations, the AONs designed to target the downstream PEs were effective and for each variant at least one AON showed high or complete rescue of the defect. Ideally, we aimed at designing one or more molecules that would efficiently correct all intron 13 splicing defects, thus permitting their use in larger cohorts of patients carrying different variants in the same hotspot. The two best-performing AONs are AON-B and AON-C, both of which proved efficacious for 3/4 of the tested variants. AON-C, targeting the dual splice site shared by PE3 and PE4, was the most efficacious for the initial set of three variants selected for the study, but had no effect on the newly identified c.1938-724A>G, as it falls completely outside of the shorter PE2 sequence. In addition, it should be noted that AON-C is a variant-specific AON for c.1938-621G>A. Consequently, AON-C has one mismatch with the mutant sequence carrying the other variants. Nevertheless, in this study, the AON was able to prevent PE3 inclusion, which might be because of the weak destabilization strength of the mismatch, although the artificial overexpression system of the midigene splice assay might also play a role. Despite leading to the same splicing defect, c.1938-619A>G shows little to no residual correct transcript and is categorized as a severe variant, whereas c.1938-621G>A is categorized as mild/benign with ∼80% of the correct transcript. As classical STGD1 is caused by combinations of severe and mild variants or two moderately severe variants, patients carrying c.1938-621G>A will most likely harbor null or severe variants on the other allele. Thus, even complete rescue of the splicing defect by AON treatment is unlikely to lead to an overall therapeutic effect on individuals carrying the c.1938-621G>A variant, whereas splicing restoration in patients with the severe variant c.1938-619A>G overall will be more impactful. Regardless of the complexity of designing an AON on the dual splice site, this was further confirmed to be an optimal target by the fact that AON-C was also able to reach ∼80% rescue of PE4, caused by the severe variant c.1938-514A>G. AON-B efficiently prevented the insertion of both PE3 and the novel PE2, thus resulting in a promising therapeutic molecule for 3/4 tested variants. Remarkably, it also reached ∼20% rescue for c.1938-514A>G, despite targeting an upstream cryptic splice site. Overall, intron 13 is a hotspot region for DI variants where novel pathogenic variants are likely to be identified in the future and the AONs designed to target the region harboring the known PEs have the potential to efficiently rescue yet undescribed variants.
AON rescue was also effective for c.6148-84A>T. This intron 44 variant was previously described to lead to three different missplicing events, i.e., PE5 insertion in combination with exon 44 skipping, PE6 insertion, and skipping of exon 45. In-depth analysis of the aberrant transcripts obtained from the midigene splice assay in this study revealed additional defects that were not previously described. In particular, PE5 insertion was, in its majority, present on its own and only in a small fraction containing exon 44 skipping, and a novel PE (PE7), which was only 20 nt shorter than PE6 was identified. Interestingly, in silico reanalysis of the variant with SpliceAI (Supplementary Table S4B) revealed that all three PEs correspond to predicted SAS strength gains, supporting the validation of the splicing defect in this study. Despite this, treatment with the designed AONs proved effective and, in particular, AON-F showed almost complete prevention of aberrant splicing.
The newly added complexity of the intron 44 variant splicing defect highlights how further studies are needed in more complex systems, more closely recapitulating the genetic environment of the retina, such as PPCs or retinal organoids. While the HEK293T-based midigene assays are a versatile platform whose main strengths lay in the capability of enabling the screening of many variants and AONs in a cost-effective way, tissue-specific transcription factors, and a larger genomic context will likely improve the overall understanding of complex splicing defects and aid in the evaluation of the effectiveness of AON molecules. In addition, the next essential steps toward clinical implementation of the AONs require adequate cell and animal models to conduct a detailed safety and toxicology assessment. Another challenge to consider is the small number of patients who would benefit from the described AONs, despite the identification of multiple variants leading to the same or similar spicing. Nevertheless, trials with N-of-1 or N-of-few have shown promising results in other rare diseases. 40 Considering the presence of rare and ultrarare splice-altering variants in ABCA4, personalized genetic treatments hold much potential for STGD1 patients.
In conclusion, AON-based splicing modulation is a promising approach to target complex splicing defects in ABCA4. In this study, we show that a single AON can efficiently target similar splicing defects localized in hotspot regions and caused by the same or different pathogenic variants. In addition, we show indirect evidence that recursive splicing is involved in the regulation of intron 13 of ABCA4.
Footnotes
Acknowledgments
The authors would like to thank Elise Heon for ascertaining a person with STGD1.
Author Disclosure Statement
R.W.J.C is the Chief Scientific Officer of Astherna B.V.
Funding Information
This work was supported by the RetinaUK, grant no. GR591 (to F.P.M.C.), a Horizon 2020, Marie Sklodowska-Curie Innovative Training Network entitled European Training Network to Diagnose, Understand and Treat Stargardt Disease, a Frequent Inherited Blinding Disorder-StarT (813490) (to R.W.J.C., F.P.M.C.), the Foundation Fighting Blindness USA, grant no. PPA-0517-0717-RAD (to A.G., R.W.J.C., F.P.M.C.), the Rotterdamse Stichting Blindenbelangen, the Stichting Blindenhulp, and the Stichting tot Verbetering van het Lot der Blinden (to F.P.M.C.), and by the Landelijke Stichting voor Blinden en Slechtzienden, the Macula Degeneratie fonds and the Stichting Blinden-Penning that contributed through Uitzicht 2016-12 (to F.P.M.C.). This work was also supported by the ProRetina Foundation Germany, the Stichting Blindenhulp, the Stichting ter Verbetering van het Lot der Blinden, the Gelderse Blindenstichting, the Stichting voor Ooglijders, and the Oogfonds that contributed through Uitzicht 2020-17 (to F.P.M.C.). This work was also supported by EJPRD19-234 (Solve-RET) (to F.P.M.C.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.