
Abstract
This study investigates the influence of heat stress preconditioning on cognitive outcome for rats with diffuse axonal injury (DAI), and attempts to examine the underlying mechanisms. Wistar rats were divided into four groups: rats subjected to heat stress preconditioning 24 h before induction of DAI (n = 10; HSDAI group), a DAI alone group (n = 10), a heat stress alone group (n = 10), and a sham-injury group (n = 10). From day 14 post-injury, the rats' learning abilities and memory were tested using the Morris water maze (MWM) task, followed by long-term potentiation (LTP) recording of the hippocampus. In addition, hematoxylin and eosin staining (H&E) and immunohistochemical staining (IHC) were conducted to determine the presence of brain lesions and expression of heat shock protein 70 (HSP70) at 24 h, and on days 14 and 20 post-injury. The rats in the DAI group displayed impaired MWM performance and attenuated LTP compared to the sham group (p < 0.05); the rats in the HSDAI and HS groups showed significant improvement in both MWM and LTP compared with the DAI group (p < 0.05), and no significant differences with the sham group (p > 0.05). Following injury, retraction balls, shrunken neurons, and HSP70 expression were visible in the brains of rats from the DAI and HSDAI groups; recovery was expedited in the rats belonging to the HSDAI group, as these pathological changes were alleviated, coincident with higher expression of HSP70. The rats' abilities for learning and memory were impaired following DAI; this may be due to the disconnection of brain regions, damage to neurons in the hippocampus, and a decrease in synaptic plasticity. Heat stress preconditioning is able to significantly attenuate this cognitive impairment, possibly mediated by the neuroprotective effect of HSP70.
Introduction
D
In the central nervous system (CNS), cells subjected to heat stress produce a group of proteins called heat shock proteins (HSPs). Once HSPs are induced, neurons show a marked resistance to subsequent stress such as ischemia, hyperthermia, and trauma (Lin et al., 2004). HSPs belong to five main families, of which the major family comprising the highly inducible HSP70 is of particular significance to the CNS (Hung et al., 2004). According to recent studies, HSP70 induced by heat stress preconditioning significantly ameliorated the pathological changes of injured axons. The studies suggested that the expression of HSP70 was enhanced and prolonged by the preconditioning, rendering it more effective in exerting its neuroprotective role and facilitating cell survival during subsequent injury (Li et al., 2005; Kelty et al., 2002).
The period of impaired learning and poor memory following DAI may persist for years. The pathological basis of this cognitive deficit, and the effect of heat stress preconditioning on it, remain to be elucidated. In this we used behavioral, electrophysiological, and morphological methods to address these questions, and to explore the underlying mechanisms.
Methods
Experimental groups
Female Wistar rats (specific pathogen free, weighing 250 ± 25 g, obtained from the Chinese Academy of Medical Sciences) were randomly assigned to four groups: an HSDAI group (n = 10; rats receiving heat stress preconditioning 24 h before induction of DAI), a DAI group (n = 10; rats subjected to DAI as injury controls), an HS group (n = 10; rats subjected to heat stress as stress controls), and a sham group (n = 10; rats sham-operated without DAI or heat stress). All experiments were carried out in accordance with the principles in the NIH guide for the Care and Use of Laboratory Animals, and were approved by the Animal Care Committee at the Animal Center, Chinese Academy of Sciences, Shanghai, China.
Heat stress protocol
Each rat was anesthetized with 10% chloral hydrate (0.3 g/kg IP) and wrapped in a temperature-controlled heating pad until its body temperature, as monitored by a rectal thermometer, reached 42°C. This condition was then maintained for 15 min (Lin et al., 2004). To prevent severe dehydration associated with hyperthermia, a 5-mL bolus of normal saline was given IP when the rat's temperature reached 39°C, 42°C, and at the end of the hyperthermic phase. The rat was then allowed to recover at room temperature with food and water ad libitum.
Induction of diffuse axonal injury
The rat model of DAl was generated using the impact-acceleration method (Morales et al., 2005). The rat was anesthetized with 10% chloral hydrate (0.3 g/kg IP). A midline incision was performed to expose the skull between the coronal and lambdoid sutures. Using dental cement, a metallic disc (18 mm in diameter and 4 mm in height) was firmly fixed to the central portion of the calvarium. Then the animal was placed in prone position on a foam bed of known spring constant under a vertical PVC tube (1.5 m in height), with the metallic disc centered under the lower end of the tube. A 500-g weight was allowed to fall through the tube from 1.5 m high to impact the disc. After the impact, the injured animal was immediately removed from the device to prevent rebound impact, and respiration and consciousness were monitored. Once the rat regained normal breathing the disc was gently removed. After examination for the presence of skull fracture, the scalp was sutured. No resuscitation was performed and the mortality rate was assessed at 24 h post-injury. Sham-injured rats were subjected to the same protocol; however, before the actual induction of injury, the disc was removed and the scalp sutured.
Morris water maze task
The water maze consisted of a 1.5-m-diameter circular tank divided into four equal quadrants (I, II, III, and IV), and a 10-cm-diameter platform was submerged 2 cm below the water surface in the center of quadrant III. The water was made opaque using nontoxic black ink and maintained at 25 ± 1°C. Movement of rats in the maze was monitored by a CCD camera connected to a personal computer, where data were collected and analyzed. The task consisted of two phases: place navigation and spatial probe. Initially, in place navigation rats were subjected to two sessions of four trials per day for five consecutive days covering days 14–18 post-injury. In each trial, the rats were released into the water individually from one of four starting points spaced 90° apart around the perimeter of the tank. They were allowed to swim freely until they reached and stayed on the platform. If they failed to locate the platform within 120 sec, they were placed on it for 15 sec. The time required to find the platform (escape latency) and the swimming speed were recorded. Subsequent starting points proceeded in a clockwise manner for the ensuing trials. There were intervals lasting approximately 5 min between trials, and 5 h between the two sessions. For the second phase, the spatial probe on day 19 post-injury, the platform was removed from the tank. The rats were released individually into water from the starting point of quadrant I and allowed to swim for 120 sec. The time spent (time percentage) in the target quadrant (quadrant III), and the number of platform location crossings were measured. There were two sessions spaced at an interval of 5 h in this phase.
Long-term potentiation recording
On day 20 post-injury, each rat was anesthetized with 30% urethane (1.2 g/kg IP) and placed in a stereotaxic instrument (Narishige, Tokyo, Japan) with its head fixed. Under a surgical microscope, the surface of the skull was exposed and two tiny holes were made in the left side with a dental drill. Incisions were made in the dura mater. Stimulating electrodes (Advent Co., Halesworth, U.K.) were placed in the perforant path (PP; location: AP −7.8 mm, lat 4.2 mm, DV 2.8 mm). A recording electrode (Advent Co.) was positioned in the granule cell molecular layer of the dentate gyrus (DG; location: AP −3.4 mm, lat 2.0 mm, DV 3.0 mm). Both electrodes were adjusted to trigger an optimal excitatory postsynaptic potential (EPSP) produced by the granule cells of the DG in response to stimulation from PP afferents. Once the response stabilized, it was recorded under low-frequency stimulations (0.1 Hz) for 15 min as the baseline. Subsequently, tetanic stimulation (10 pulses at 100 Hz for 5 msec repeated five times) was delivered to induce the LTP. The EPSPs were amplified by a conventional amplifier (AD Instruments Pty. Ltd., Castle Hill, Australia) at a frequency band of 5 Hz–5 kHz, and recorded six times respectively, at the time points of 0, 10, 20, 30, 40, 50, and 60 min following tetanic stimulation. All EPSPs were monitored on an oscilloscope and digitized at a sampling interval of 20 μsec for on-line computer display. The LTP was measured using Chart v5.3 and expressed as percentage changes of maximal initial EPSP slopes from mean response of the baseline: (Slopeafter tetanus/Slopemean of baseline) × 100%.
Histology and immunohistochemistry
At 24 h, and on days 14 and 20 post-injury, two rats from each group were anesthetized with 30% urethane (1.2 g/kg IP) and transcardially perfused with 0.01 M phosphate-buffered saline (PBS), followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB) (pH 7.0). After that, the brains were removed and immersed in the same fixative at 4°C for at least 24 h, and then were embedded in paraffin for tissue sectioning. Coronal 5-μm sections comprising the hippocampal area, and horizontal 5-μm sections of the brainstem were cut using a rotary microtome. The sections were used for hematoxylin and eosin (H&E) staining in accordance with the standard procedure, and immunohistochemical staining for β-amyloid precursor protein (β-APP, a marker of DAI), and HSP70 using the avidin-biotin complex procedure as follows.
After deparaffinization and hydration, the sections were treated with 0.5% Triton X-100 for 10 min and then incubated in 3% hydrogen peroxide for another 10 min. For antigen retrieval, the sections were heated in 0.01 M citrate buffer (pH 6.0) at 121°C for 3 min using an autoclave. Next the sections were immersed in 10% normal goat serum for 10 min, and incubated overnight with rabbit anti-β-APP polyclonal antibody (1:100; Boster, Wuhan, China) at 4°C. On the following day, the sections were incubated with biotinylated goat anti-rabbit IgG at 25°C for 10 min in a vacuum oven (−0.06 MPa), followed by incubation with avidin-biotin peroxidase complex under the same conditions for 10 min. Finally, the immunocomplex was visualized with the aid of the chromogen diaminobenzidine. A duplicate set of slides was stained similarly, with PBS replacing β-APP antibody, and served as the negative control. After dehydration, the slides were mounted and examined under a microscope (Leica DM RXA2; Leica, Solms, Germany), and images were captured using a digital camera (Leica 300F). IHC staining for HSP70 was also conducted following the same procedure, and 10% rabbit serum, mouse anti-HSP70 monoclonal antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), and biotinylated rabbit anti-mouse IgG were used in the relevant steps. Axonal retraction balls (ARBs, evidence of axon disruption) and immunoreactive cells were counted by two independent examiners, within five stochastic high-power fields in five regions (cerebral cortex, subcortical white matter, corpus callosum, hippocampus, and brainstem) of each brain.
Statistical analysis
All data are presented as mean ± SEM. Escape latencies and swimming speeds in place navigation were compared using a two-way ANOVA for repeated measures. Data for spatial probe, LTP recording, H&E staining, and IHC staining were compared using a one-way ANOVA. Significant differences between means were determined by Duncan's multiple range tests with p < 0.05 set as the level of statistical significance.
Results
Acute neurologic responses
The impact resulted in brief apnea (0–20 sec) and/or dyspnea (0–3 min) with or without generalized convulsions, followed by lasting periods of coma (manifested as loss of corneal reflex and delayed recovery of consciousness). Respiration gradually returned to normal within 15 min, and no skull fractures were found. Recovery of consciousness was 139.65 ± 5.36 min in the DAI group, and 124 ± 4.94 min in the HSDAI group, but no significant difference was found between them (p > 0.05). Rats in the sham group regained consciousness in 99.64 ± 6.75 min, which differed significantly from the former two groups (p < 0.01). Rats that did not survive experienced apnea lasting for up to 20 sec, or dyspnea lasting up to 3 min. The cause of death was central respiratory depression. Mortality rates at 24 h were 36.84% (7 out of 19 rats) in the DAI group, and 23.53% (4 out of 17 rats) in the HSDAI group. Despite the similarities in neurological response to injury, the mortality rates were vastly different; based on these observations we infer that to some extent, rats with heat stress preconditioning appeared to be more resistant to impact-induced acute neurological changes.
Morris water maze performance
During place navigation, the performance of all groups improved with training. After circling the periphery of the tank for the first few trials, rats exhibited increasingly efficient exploratory strategies as the trials continued, until they succeeded in locating the platform quickly. Therefore the average escape latencies on successive days decreased remarkably from day 1 to day 3, and then stabilized for the subsequent days (Fig. 1). This was proven by a two-way repeated-measures ANOVA that showed a significant effect of time (i.e., the day under evaluation) (F = 78.42, p < 0.0001), and the Duncan's test that revealed the learning pattern. There was also a significant effect of treatment (i.e., by group) (F = 17.17, p < 0.0001), for which the Duncan's test revealed that latencies were prolonged significantly in the DAI group compared with those of the HSDAI, HS, or sham group on each training day (p < 0.05; Fig. 1). This suggested that it took DAI rats more time to find the platform. In sharp contrast, HSDAI rats mastered the task rapidly and located the platform with shorter latencies, demonstrating superior spatial learning compared to DAI rats. Escape latencies of the HSDAI group did not differ significantly from those of the sham or HS group (p > 0.05; Fig. 1). In addition, the swimming speeds for each group remained constant throughout testing, with no significant differences seen among groups (p > 0.05; Fig. 2). This suggests that motor function was not the underlying determinant for the prolonged latencies. Data in this phase demonstrate that learning acquisition was impaired in DAI rats, but heat stress preconditioning effectively prevented this impairment, and heat stress per se did not interfere with acquisition performance.

Escape latency in place navigation. Note that the latencies of the HSDAI, HS, and sham groups were all significantly shorter than those of the DAI group (p < 0.05), and there were no significant differences among the HSDAI, HS, and sham groups (p > 0.05; *p < 0.05 for the DAI group versus the other groups on a specific day).

Swimming speed in place navigation. No differences were observed among the four groups throughout testing (p > 0.05).
In the spatial probe, one-way ANOVA demonstrated a significant effect of treatment, in both percentage of time spent in the target quadrant (F = 3.43, p < 0.05), and number of platform location crossings (F = 5.41, p < 0.01). Duncan's test revealed that DAI rats spent significantly less time in quadrant III, and crossed the platform location less frequently than did HSDAI, HS, or sham animals (p < 0.05); no differences were found among the latter three groups (p > 0.05, Figs. 3 and 4). This indicates that memory retention was impaired by DAI, protected by heat stress preconditioning, and not affected by heat stress alone.

Time percentage in the target quadrant during spatial probe testing. Note that preference for the target quadrant for DAI rats was significantly lower than that of the HSDAI, HS, or sham rats (p < 0.05). No differences were found among the latter three groups (p > 0.05; *p < 0.05 for the DAI group versus the other groups).

Number of platform location crossings during spatial probe testing. Note that DAI rats crossed the platform location less frequently than HSDAI, HS, or sham rats (p < 0.05). No differences were found among the latter three groups (p > 0.05; *p < 0.05 for the DAI group versus the other groups).
Long-term potentiation
One-way ANOVA revealed a significant effect of treatment (F = 89.54, p < 0.0001). Duncan's test showed significant differences in LTP between the DAI group and the sham group (p < 0.05), as well as between the DAI group and the HSDAI group (p < 0.05); however, no differences were found between the HSDAI group and the sham group (p > 0.05), or between the HS group and the sham group (p > 0.05).
This finding indicates that the tetanic stimulation induced significantly greater increases in EPSP slopes in the sham group than in the DAI group (Fig. 5A). LTP in the DG was severely inhibited in DAI rats. However, heat stress preconditioning successfully prevented this LTP suppression, as is evident from the fact that the increase in EPSP slopes in HSDAI rats was more significant than that of the DAI rats (Fig. 5B), and this LTP approached the level of sham controls (Fig. 5C). Although following tetanus, the increase in LTP for the HSDAI rats was not as rapid as in the sham rats, a marked protective effect of heat stress preconditioning against injury-induced LTP attenuation was evident. In addition, the HS group displayed no significant difference in the increase of EPSP slopes compared with the sham group (Fig. 5D), suggesting that heat stress per se had no detrimental effect on LTP in the DG.
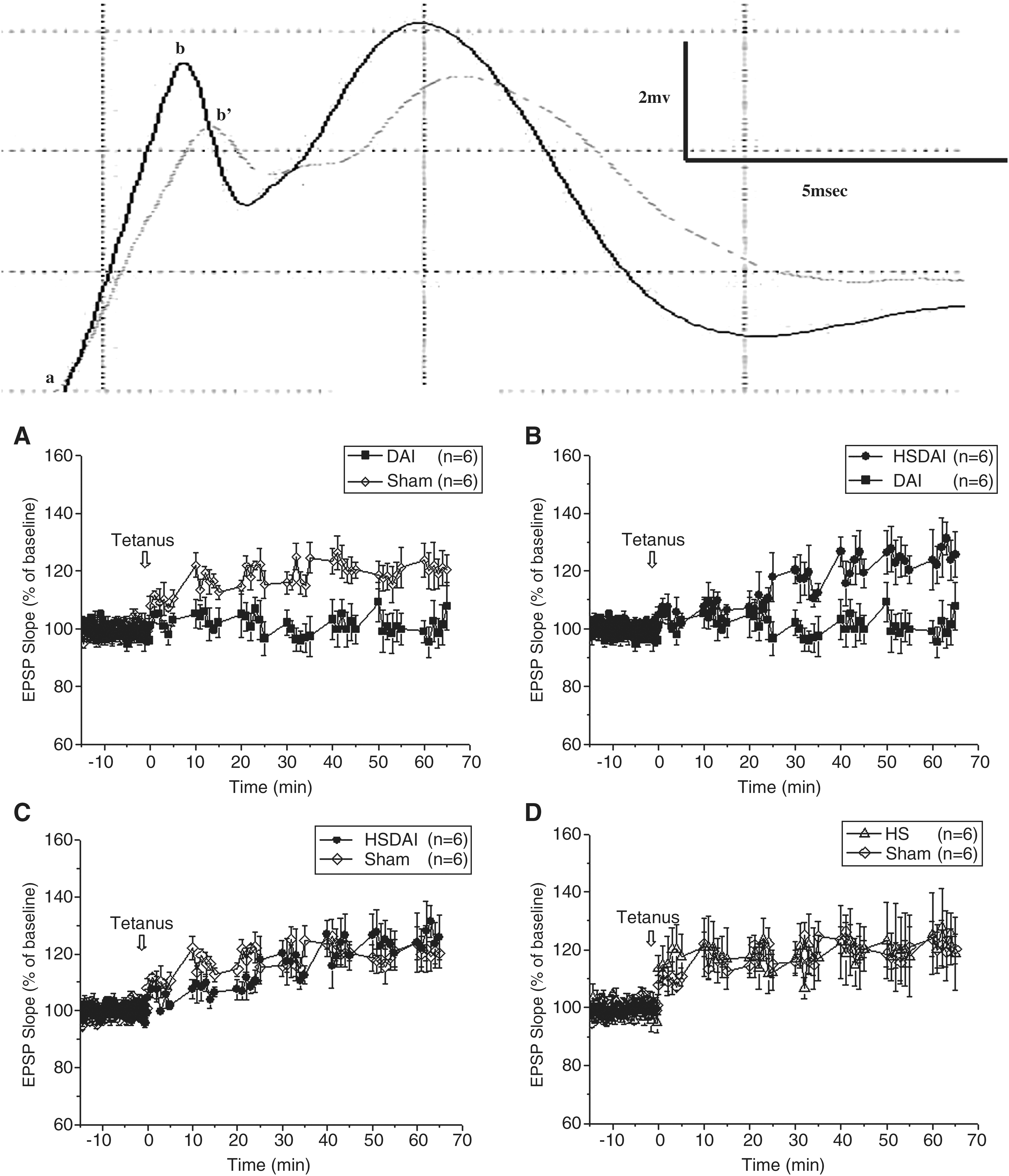
Sample trace and changes in EPSP slopes after tetanic stimulation (arrows). The slope was measured from a to b' in the trace of baseline (dashed line), and from a to b in the trace after tetanus (solid line). (
Histopathology
Macroscopic examination of the brains showed an absence of focal lesions or contusions. Subarachnoid hemorrhage was usually observed in the basal cistern, cisterna magna, and the surface of the cerebral hemisphere. In rats that did not survive, there were obvious hemorrhagic lesions in the brainstem.
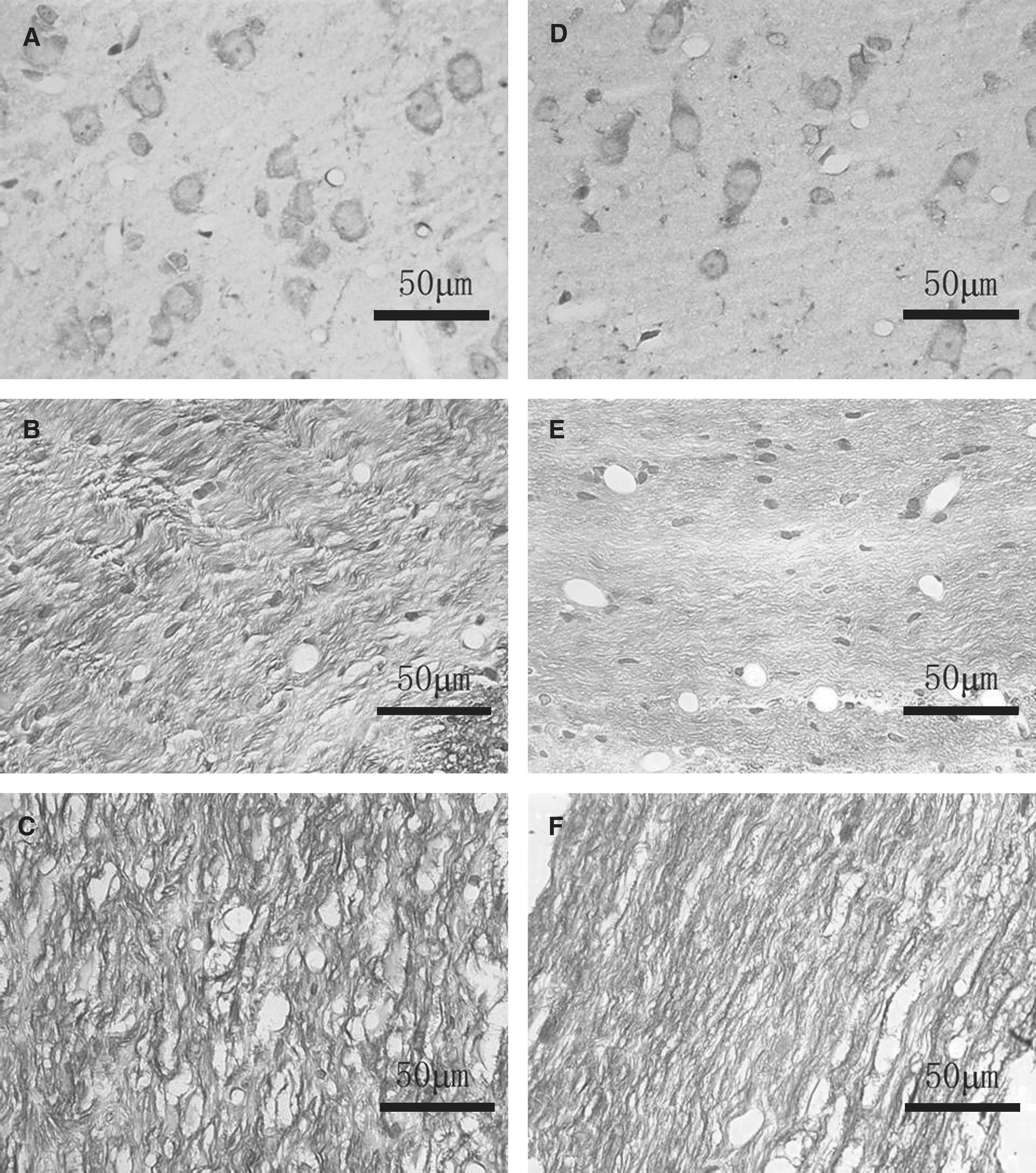
In the DAI group at 24 h post-injury, H&E staining revealed round/oval eosinophilic ARBs (Fig. 6A) scattered in the cerebral cortex, subcortical white matter, corpus callosum, and brainstem. There were diffuse swollen axons in wave-like form with ARBs connected to or disconnected from them (Fig. 6A and B). In the cerebral cortex and hippocampus, pink shrunken neurons were associated with perineuronal vacuolations, indicating neuronal degeneration and death. There were reduced neuronal density and disrupted cellular alignment in the hippocampal CA3 area (Fig. 6C). Also, red blood cells congested engorged capillaries (Fig. 6A), and the subarachnoid space. At day 14, though smaller in size and number, ARBs and axonal varicosities were still visible, together with degenerated neurons and vascular congestion, in the cerebral cortex and hippocampus. At day 20, there were local axonal varicosities and a few degenerated neurons left. In the HSDAI group at 24 h, there were fewer ARBs in the aforementioned brain regions, compared to the DAI group (5.02 ± 1.54/high-power field [hpf] versus 9.28 ± 3.33/hpf; p < 0.001; Fig. 6D). The swollen axons also appeared more organized (Fig. 6E). While degenerated neurons and congested capillaries were also noted in the cerebral cortex and hippocampus, cellular density and alignment in the CA3 area were relatively normal (Fig. 6F). By day 14, there were local axonal varicosities and hardly any ARBs. The density of degenerated neurons lessened, while regeneration was initiated for most of the injured axons. By day 20, the axons appeared normal, without visible ARBs, and only a few degenerated neurons were visible. Comparable sections from the HS and sham groups appeared normal at all time points.

H&E staining at 24 h post-injury. (
IHC staining for β-APP revealed positive neurons, with brown granules in the cytoplasm and axon, distributed throughout the cerebral cortex and hippocampus in the DAI group at 24 h post-injury (Fig. 7A). Injured axons, characterized by swelling, distortion, and disconnection from ARBs, containing accumulated β-APP, were present in the subcortical white matter, corpus callosum, and brainstem (Fig. 7B and C). The intensity of the immunoreaction in those axons varied segmentally. At day 14, the density of β-APP-positive neurons decreased, but these cells were still clustered in some areas such as the DG. Injured axons were still visible, but to a lesser extent. By day 20, mild immunoreactivity was seen in a few neurons, with no staining of axons. Immunostaining in the HSDAI group at 24 h was moderate; there were fewer positive neurons in the aforementioned areas compared with the DAI group (9.86 ± 1.82/hpf versus 12.28 ± 3.50/hpf; p < 0.05; Fig. 7D), and less axonal changes in the corpus callosum and brainstem (Fig. 7E and F). There were fewer β-APP-positive neurons and lighter-stained axons on day 14, with no visible immunostaining of axons on day 20. In the HS group mild immunoreactivity was present in a few neurons in the cerebral cortex at 24 h.
IHC staining for β-APP at 24 h post-injury. (
IHC staining for HSP70 at 24 h revealed brown granules within the nuclei of neurons and glial cells in the DAI group; HSP70-positive cells were accumulated in the corpus callosum, thalamus, hippocampus (Fig. 8B and C), and medial lemniscus. A few scattered positive cells were visible in the cerebral cortex (Fig. 8A) and brainstem. In the HSDAI group, HSP70-positive cells were distributed in the same regions as in the DAI group (Fig. 8D–F); however, they were more numerous (32.12 ± 6.58/hpf versus 18.66 ± 3.03/hpf; p < 0.001), and had intense staining, especially in the hippocampal fimbria (Fig. 8F). This indicates higher expression of HSP70. In the HS group, mild immunoreactiviy was present in the cerebral cortex, brainstem, hippocampus, and corpus callosum. There were no stained cells visible in any group by days 14 and 20.

IHC staining for HSP70 at 24 h post-injury. (
Discussion
In this study, we present evidence for widespread damage to the axons and neurons in the cerebral cortex, subcortical white matter, corpus callosum, hippocampus, and brainstem of injured rats. These results are in agreement with results presented in a previous study (Meythaler et al., 2001), and are similar to the pathological features seen in human DAI. In addition, some of the most extreme conditions experienced by DAI victims (i.e., accelerating force on the brain caused by an impact to the closed skull, respiratory depression, and the reduced ventilation seen immediately after the injury) were successfully reproduced by this model.
MWM is a well-validated method for evaluating learning and memory in rodents. In our study, the significant underperformance on the MWM task at 2 weeks post-injury suggests that rats with DAI experience chronic disruption of learning and memory. This is in accordance with earlier reports suggesting that acquisition and retention capacities in rats are impaired in this DAI model, and that the rats are unable to fully recover until 11 days post-injury (Morales et al., 2005; Cernak et al., 2004). This behavioral outcome is also supported by histological and electrophysiological analyses.
We propose that the cognitive impairment may be primarily due to three reasons: disconnection of brain regions, damage to neurons in the hippocampus, and a decrease in synaptic plasticity. First, a rat's performance on the MWM depends on the coordinated interaction of different brain regions that constitute a functionally integrated neural network. The integrity of the connections among brain regions is necessary for spatial learning and memory (Lynch, 2004). DAI leads to massive destruction of white matter tracts and destroys those connections. Therefore, chronic cognitive impairment emerges during the slow recovery process of injured axons. Second, the hippocampus is necessary for acquisition, retrieval, and consolidation of spatial information (D'Hooge and De Deyn, 2001). The pathological changes we observed in the hippocampus could be related to the impaired performance on the MWM task, because hippocampal lesions would lead to a poor ability to localize and navigate (as manifested by extended searching) (Leggio et al., 2006). Damage to the hippocampus in DAI may be due to the following factors, including its vulnerability to injury, in which glutamate release and increased intracellular calcium may trigger a cytotoxic cascade (D'Hooge and De Deyn, 2001). In addition, the hippocampus is positioned in the temporal lobe at the end of long fiber tracts, and perisomatic axonal injury would immediately translate into ultra-rapid cell death (Buki and Povlishock, 2006). Although some injured neurons might survive, it would take weeks for them to restore normal function. Third, LTP in the hippocampus is generally accepted as the cellular foundation for learning and memory (Miyamoto, 2006). The two roles of the hippocampus in solving the MWM task, self-localization and route replay, depend on the LTP process. Saturating the LTP in the DG impairs MWM performance (D'Hooge and De Deyn, 2001). In the current study, the impaired MWM performance demonstrated by the DAI group could be related to the suppression of LTP. Furthermore, LTP is involved in behaviorally-relevant synaptic modification that contributes to spatial cognition (Lynch, 2004). Reconstruction of synapses is essential in developing LTP and memory following injury (Miyamoto, 2006); however, the injury may be too severe for the brain to generate appropriate reconstructive activity (Buki and Povlishock, 2006). In our study, the cognitive deficits induced by DAI persisted for 2–3 weeks, which could be the result of damaged or inappropriate projections from the PP to the DG, or other maladaptive changes that decease synaptic plasticity.
Despite the obviously inferior performance on the MWM task, DAI rats still displayed spontaneous learning and a certain extent of memory, whereas their EPSP slopes remained essentially unchanged after tetanic stimulation in LTP recording. These results seem inconsistent, but they can be explained this way: one possibility is that although the LTP of the PP to the DG is impaired in DAI rats, the LTP of other synaptic connections, such as the DG to CA3 and/or CA3 to CA1, may have affected the rats' abilities in solving the MWM task. However, those connections may not be intact either, leading to a decrease in overall synaptic plasticity, and thus poor spatial cognition on the MWM task. Another possibility is that other cellular mechanisms underpinning learning and memory may exist in the hippocampus, and may serve to compensate LTP impairment. This is supported by some studies showing that MWM learning was still possible in animals when LTP processes were blocked (D'Hooge and De Deyn, 2001; Myhrer, 2003; Leggio et al., 2006).
To the best of our knowledge, this is the first work to demonstrate that heat stress preconditioning protects rats with DAI from secondary impairment of cognitive function. The preconditioning also leads to improved survival post-injury and reductions in histopathological sequelae. The neuroprotective effect may be mediated by HSP70, which is highly expressed in the HSDAI group, especially at 24 h after heat stress, the time point at which DAI was induced. On the other hand, although HSP70 induction was also observed in the DAI group, the protection afforded was insufficient due to its relatively low expression. Possible mechanisms underlying this protection are discussed below.
Heat stress preconditioning, possibly mediated by HSP70, may provide fundamental protection of axonal and neuronal function. As a molecular chaperone, HSP70 limits the damage to cells by preventing protein aggregation, eliminating denatured proteins and regulating both protein translocation and protein folding (Beere and Green, 2001). Restoration of normal function in injured cells may therefore be promoted via several potential mechanisms. First, by combining with the cytoskeletal proteins, HSP70 protects them from enzymolytic degradation by caspases, and participates in the formation and restoration of microtubules (Su et al., 2008). This is essential for regulating axonal projections and synaptic modification. It also creates optimal conditions for the sprouting of adjacent nerve fibers, and leads to the recovery of synaptic input to the previously deafferentated neurons. Functional connections are restored through this mechanism, and constructive communication among brain regions can be reinstated. Second, HSP70 not only limits glutamate damage to neurons in the CA1, CA3, and DG of the hippocampus (Sato and Matsuki, 2002), it also blocks excessive calcium influx by combining with transport proteins to restore their function (Su et al., 2008). In addition, HSP70 suppresses post-injury apoptosis and facilitates cell survival by inhibiting cytochrome c release and caspase activation, and by modulating the pro- and anti-apoptotic members of the Bcl-2 family (Beere and Green, 2001). Third, both heat stress preconditioning and incubation with HSP70 solution protect synaptic transmission and help maintain normal function and plasticity in hippocampal synapses (Kelty et al., 2002). At the presynaptic terminal, the complex of HSP70 and cysteine string protein contributes to synaptic plasticity via modulation of neurotransmitter release (Tobaben et al., 2001). Moderate levels of glutamate and calcium play an essential role in the induction of LTP (Miyamoto, 2006), facilitating learning acquisition and memory retention; however, excessive glutamate release and calcium overload have the opposite effect (D'Hooge and De Deyn, 2001), and lead to secondary axotomy in DAI. HSP70 helps in moderating the release of these neurotransmitters to optimally regulated levels. In the postsynaptic membrane, HSPs, including HSP70, activate protein kinase C or mitogen-activated protein kinase, both of which are essential for the induction and maintenance of LTP (Lin et al., 2004). In addition, the AMPA receptor, which is necessary for LTP expression, is also upregulated (Okada et al., 2003; Gerges et al., 2004). Enhanced activity of the AMPA receptor in the DG has been shown to improve learning and memory (Okada et al., 2003). These mechanisms, which cover the three main reasons for cognitive impairment following DAI, may underlie the improvement in outcomes for the MWM task and LTP recordings for the HSDAI group. On the contrary, without sufficient protection from HSP70, increased damage to relevant structures and more severe functional impairment were observed in the DAI group.
Other functions of HSP70 following heat stress preconditioning may also contribute to its neuroprotective role. For example, HSP70 may prevent excitatory amino acids from interacting with their receptors (Kelty et al., 2002). Another important function is related to the accumulation of β-APP in DAI. β-APP is enzymatically lysed into the neurotoxic β-amyloid, which has been linked to impaired spatial learning and memory, and additionally to deficiencies in LTP (Brody and Holtzman, 2006; Lynch, 2004). HSP70 may diminish the aggregation of β-APP, prevent the enzymolytic process, and weed out some of the irreversible β-amyloid. However, we cannot eliminate the possibility that heat stress preconditioning exerts neuroprotection by orchestrating changes in the expression of other proteins, either independently or in combination with HSP70. This possibility will be investigated in our future studies.
Investigation of cognitive outcomes in rats may provide insight into cognitive recovery in humans. Spatial cognition is an advanced brain function for rats, and hence provides a valuable model. The three reasons for impairment discussed above may also be responsible for a significant proportion of the cognitive deficits exhibited by DAI patients. To date, there is no pharmaceutical intervention for DAI with proven efficacy (Morales et al., 2005; Li et al., 2008). A novel approach may be developed through the enhancement of intrinsic neuroprotection (Li et al., 2005). Heat stress preconditioning and HSP70 can preserve normal physiological function, as this and other studies have shown (Lin et al., 2004; Kelty et al., 2002; Sato and Matsuki, 2002), and as such may have important therapeutic implications. Further investigation to identify the cellular and molecular basis of heat stress preconditioning and HSP70's influence on DAI should provide a conceptual framework for developing novel strategies aimed at maintaining the brain in a sustained defensive state.
Footnotes
Acknowledgments
This work was partly supported by the Municipal Science Foundation Research of Tianjin (no. 06YFJMJC09400 to Z.Y.), and the National Basic Research Program of China (no. 2007CB914803 to T.Z.). We thank Dr. Alexander Gerhard for his expert editorial review of this manuscript.
Author Disclosure Statement
No conflicting financial interests exist.