
Abstract
Spinal cord injury (SCI) remains a major challenge to neurological research. Progress in both basic and clinical research has shown that neurons and oligodendrocytes are equally susceptible to such injury. In injuries secondary to direct injury to the spinal cord, oligodendrocytes appear to be highly vulnerable to various harmful factors and eventually undergo apoptosis. Due to the loss of myelinating cells, axonal demyelination is likely to affect the neural function of surviving axons. Recently, improved understanding of the pathological changes ongoing in oligodendrocytes following injury has shown that the death of these cells plays a vital role in the demyelination of axons. Because the demise of oligodendrocytes and subsequent axonal demyelination impair the conductive capacity of surviving axons, it seems reasonable to expect that reducing oligodendrocyte death and improving axonal myelination holds potential for the treatment of SCI. In the clinical setting, such therapy may help these patients, including those with complete functional injury and those with white matter preservation. Accordingly, it appears reasonable that improving axonal myelination and the conductive capacity of surviving axons will be of great benefit in patients with mild to moderate injury of the spinal cord. We here present a review of the pathophysiology and mechanisms of oligodendrocyte death and axonal demyelination that follow injury to the spinal cord, and discuss the potential for treating them. Because cell transplantation has recently become a promising strategy for replacing lost oligodendrocytes and improving axonal myelination in SCI, we also discuss the significance of cell transplantation as a novel approach to treating SCI.
Introduction
S
Both neurons and oligodendrocytes are equally vulnerable to these damaging secondary effects of SCI (Crowe et al., 1997; Xu et al., 2004; Emery et al., 1998). During the period of secondary injury, a great many oligodendrocytes undergo apoptosis, both at the epicenter of the lesion, and at sites distant from it. The consequent loss of myelin-forming cells leads to further denudation of surviving axons and impairs their conductive capacity, markedly limiting the recovery of neural function following SCI (Hulsebosch, 2002).
With advances in knowledge about oligodendrocytes and the myelin sheath in SCI, the role of oligodendrocytes in axonal demyelination and remyelination has generated great interest among neuroscientists in this formerly obscure area of pathophysiology (Beattie et al., 2000; Warden et al., 2001; Shuman et al., 1997). As has been shown, the death of oligodendrocytes and subsequent demyelination of surviving axons contribute substantially to the deterioration of neural function in SCI (Profyris et al., 2004). However, the spontaneous remyelination of demyelinated axons is severely limited in the unfavorable environment that follows injury to the cord. It therefore seems reasonable that reducing oligodendrocyte loss and improving axonal myelination are likely to have potential as a strategy for treating SCI. Oligodendrocytes also appear to be important target cells for improving neural function in other types of injury to the CNS (Schwab, 2002; McDonald and Belegu, 2006). With regard to optimal recovery of neural function in SCI, preventing the death of myelin-producing cells and facilitating axonal remyelination are of potentially immense value to patients with incomplete injuries of the spinal cord. This article is therefore an overview of the significance of improving axonal myelination in SCI and its relevance to the functional recovery of the incompletely injured spinal cord.
The Myelin Sheath
Myelin is an essential constituent of white matter in the CNS. The myelin sheaths surrounding axons are the most abundant membrane structures in the vertebrate nervous system. The unique composition of the myelin sheath, rich in lipids and with a low water content, is responsible for its electrical insulation of axons, and the segmental structure of the sheath, which is responsible for the saltatory conduction of nerve impulses, allows it to support rapid conduction of nerve impulses along axons in the vertebrate nervous system (Morell et al., 1994). The myelin sheath has a spiral structure consisting of the plasma membranes of myelinating cells. These cells project sail-like extensions of their cytoplasmic membranes, each of which forms a segment of sheathing around an axon (Morell et al., 1994; Peters et al., 1991). The unique disposition of the segments of myelinated nerve fibers, or internodes, which is separated by unsheathed regions, brings about the so-called “saltatory conduction” that is the chief characteristic of nerve impulses conducted along myelinated tracts. Shepherd (1988) has shown that nerve impulses are generated at the nodes of Ranvier in myelinated fibers, and that the myelin sheath has a high resistance and low capacitance that make the current passing through a myelinated fiber tend to flow down the fiber to the next node, rather than leak across the membrane. Consequently, nerve impulses are able to jump from one node to the next, propagating what is called saltatory conduction.
Besides the foregoing functions of the myelin sheath, myelinating cells have other important effects on the maintenance of normal axonal function. Sanchez and colleagues (1996) have shown that signals from myelinating cells can induce axonal growth by triggering local accumulation and organization of the neurofilament network. Furthermore, oligodendrocytes have been proven capable of promoting the radial growth of axons by inducing neurofilament accumulation (Windebank et al., 1985; Waxman, 1997). Accordingly, oligodendrocyte dysfunction also has adverse effects on the normal function of axons. Griffiths and associates (1998) have demonstrated several morphological abnormalities in the axons of myelin-deficient mice, including swelling and degeneration, and particularly the presence of axonal “spheroids.” Collectively, these findings demonstrate an important role of glial-axonal communication in the maintenance of axonal integrity and function. The proper functioning of both myelin-forming cells and the myelin sheath is essential for the normal function of the CNS. Both axonal demyelination and the dysfunction of myelinating cells will significantly affect the neural function of the spinal cord.
Demyelination
Axonal demyelination is a unique feature of the pathological changes that follow injury to the nervous system. Extensive demyelination has been shown to exist in the lesioned area following SCI (Fig. 1). Lampert and Cressman (1966) demonstrated axonal demyelination of axons as a conspicuous and important pathological characteristic of SCI, and Griffiths and McCulloch (1983) described the changes in the myelin sheaths of cats with contusive SCI during the first few weeks after injury. Both partial and full-thickness demyelination of axons was frequently observed in the area of injury. These studies suggested that the myelin damage that occurs in SCI is initially mechanical, but is later aggravated by other mechanisms. Subsequently, Blight (1985) documented the morphological and pathophysiological changes in the spinal cords of cats with induced SCI. The demyelination of axons that survived the initial insult occurred chiefly between 2 and 7 days after injury. However, it has been proposed that variations in chronic demyelination at the lesion site are probably independent of the intensity of injury, and that secondary pathological mechanisms should be considered as possible contributors to the chronic deficits in neural function that follow SCI (Totoiu and Keirstead, 2005; Blight, 1992).
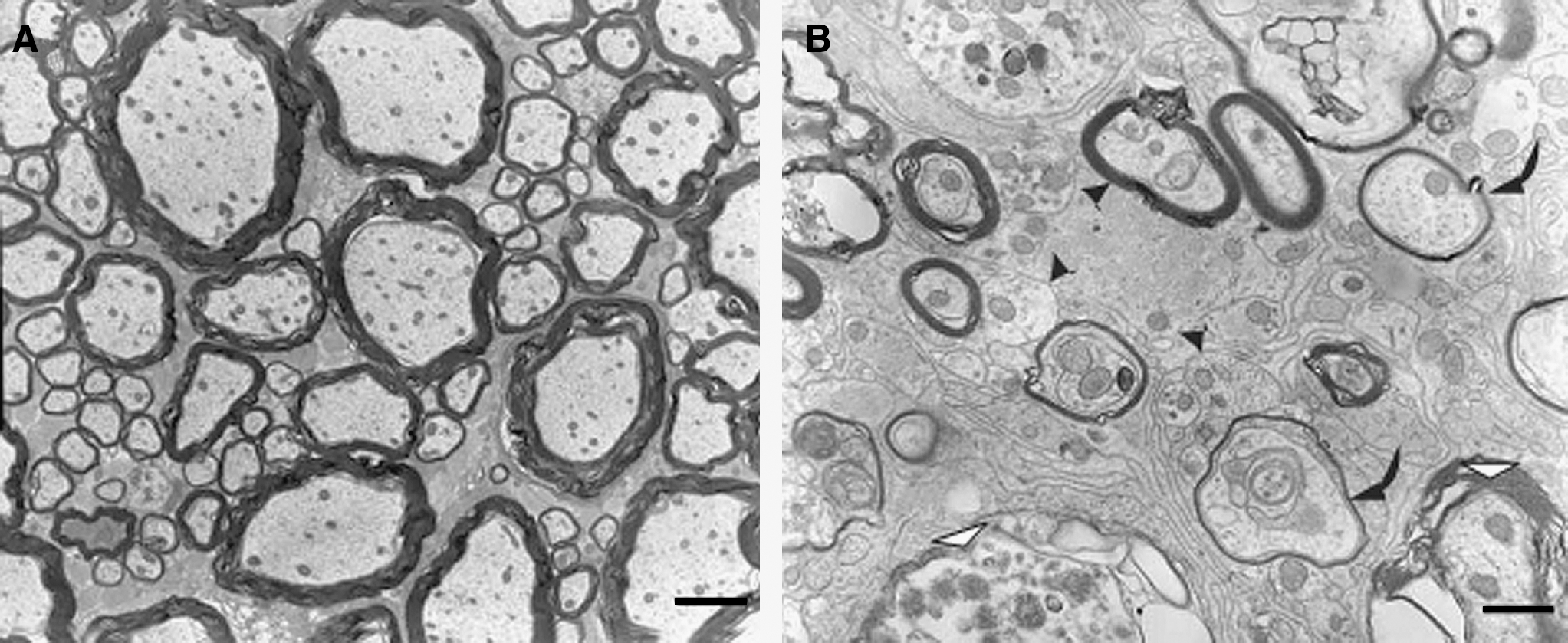
Ultrastructural analysis of axonal demyelination of injured spinal cord in experimental SCI. (
With progress in technology and further study, it has become clear that the demyelination of axons in SCI occurs initially at the epicenter of the lesion, and then progresses gradually within the fascicle of fibers that constitute the white matter of the cord. Totoiu and Keirstead (2005) performed a detailed study of the extent of axonal demyelination and its relevance to functional impairment over a period of 450 days after contusive SCI in rats. They found that the number of demyelinated axons reached a peak at 24 h and then declined from 7–14 days after injury. Axonal demyelination continued for up to 450 days after injury. Their study clearly demonstrates that demyelination of axons following SCI is a chronic pathological process, which makes it reasonable to believe that axonal demyelination has the potential to become a target in the treatment of SCI.
Besides being seen in animal models of SCI, demyelination is also prominent clinically in cases of SCI. Guest and associates (2005) presented convincing evidence for demyelination in cases of human SCI. Their autopsy study confirmed the demyelination of axons in the contusively injured human spinal cord through immunohistochemical methods. They found that axonal demyelination at the lesion site could in some cases be detected even more than a decade after injury. They also frequently observed substantial demyelination of axons in the region surrounding the injury cavity, with a rim of surviving subpial axons at the lesion site in the chronic stage of injury (Fig. 2). Thus the demyelination of axons, like the axonal pathology itself in SCI, is a prominent pathological feature of such injury in both animals and humans.
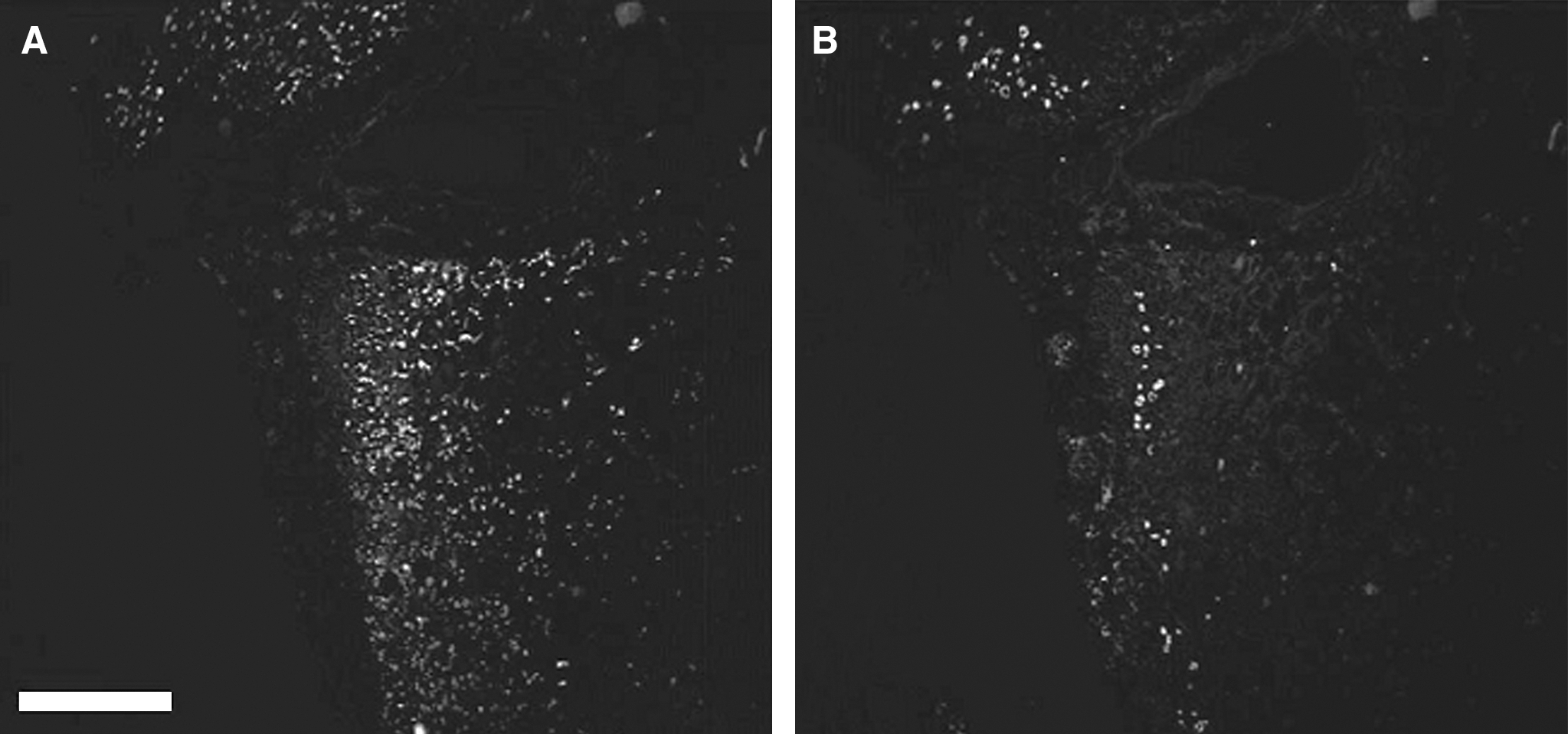
Evidence of axonal demyelination in incomplete human SCI at 1 year after injury. (

The Uninjured White Matter and Its Significance in SCI
A judicious understanding of the natural pathology of SCI is essential for experimental and clinical studies of such injury. In the clinical setting, complete anatomic transection of the dorsal column is a rare occurrence. Even in patients with SCI classified clinically as complete injury, some degree of anatomic continuity of the lesioned cord is usually preserved within the white matter. Dimitrijevic and colleagues (1983) first introduced the term “discomplete” to describe their finding of electrophysiological transmission of signals across the lesion site in patients considered clinically to have complete injuries, and who had lost all neural function below the lesion site.
Many other investigators have since studied the neurological outcomes of patients with complete cord injuries, but in whom a variable number of intact nerve fibers traversed the lesion site. A series of pathological events were found to follow the initial insult and to produce progressive secondary injuries within both the gray matter and the white matter of the cord. Besides neuronal death, marked oligodendrocyte apoptosis occurs at the lesion site and distant from the epicenter of injury (Dong et al., 2003; Beattie et al., 2000; Crowe et al., 1997; Profyris et al., 2004; Liu et al., 1997; Shuman et al., 1997; Smith and Jeffery, 2006). Despite this widespread secondary degeneration, however, varying amounts of white matter are preserved in the lesion area in most cases of SCI, down to a thin subpial rim of spared axons in cases of severe injury (Bunge et al., 1997). Unfortunately, these surviving fibers tend to become demyelinated as the consequence of an inflammatory reaction and oligodendrocyte death (Blight, 1983). This axonal demyelination is thought to be partly responsible for the long-term deficits seen in neural function following SCI.
The phenomenon of “discomplete” cord injury in SCI occurs not only in animal models, but also in humans. As noted earlier, anatomically complete injury is rare in human SCI, and injuries of lesser severity are much more common (Norenberg et al., 2004; Kakulas, 1984). Moreover, one of the most important findings in a study of 564 cases of SCI was that most of the patients retained a proportion of spinal cord white matter traversing the level of the lesion, and patients were regarded as having an “anatomically discomplete” injury (Kakulas, 1999). The finding of some anatomical continuity of white matter across the lesion site in a large proportion of patients with clinically incomplete or “discomplete” SCI strengthens the concept of axonal preservation as an approach to treating SCI.
The surviving white matter in cases of incomplete SCI would appear to have an important role in the enhancement of remyelination and other reparative strategies following SCI (Bunge, 1994). Patients with incomplete cord injuries have a better prospect for achieving functional recovery than those with anatomically complete injuries. Although the current focus of treatment of SCI is largely on the regeneration of injured axons to traverse the lesion, it seems more feasible in many cases of SCI to maximize the neural function of spared ascending and descending white matter tracts (Kakulas, 1999; Raineteau and Schwab, 2001). In both animals and humans, significant preservation of neural function of the spinal cord can be achieved if approximately 10–15% of the white matter at the lesion site is spared (Blight, 1983; Raineteau and Schwab, 2001; Helgren and Goldberger, 1993). The preservation of white matter in many cases of incomplete SCI offers a good opportunity for amplifying the residual activity of the cord, and also provides a substrate for restorative approaches to improve neural function in this clinical population. It therefore seems reasonable to achieve meaningful functional recovery in SCI either through the use of preserved white matter, or by preventing the demyelination of axons (Young, 1988).
The Effects of Demyelination on Neurological Function
Because the demyelination of axons and other pathological processes occur simultaneously in the lesioned spinal cord, it is impossible to accurately assess the extent to which axonal demyelination contributes to the impairment of neural function in SCI. However, improving neural function by preventing axonal demyelination or facilitating axonal remyelination could be considered akin to improving axonal myelination. A number of experiments have shown that such improvement is beneficial to the recovery of neural function after SCI.
The insulating property of the myelin sheath favors rapid nerve conduction. After axonal demyelination, the low density of sodium channels in the internodal axon membrane impedes nerve conduction. Furthermore, the potassium channels “unmasked” by such demyelination tend to clamp the membrane at a potential near its resting potential, further interfering with impulse conduction through demyelinated axons (Waxman, 1992). 4-Aminopyridine, a potassium channel blocking agent, has been shown to be able to improve the conduction of nerve impulses, and consequently to facilitate the recovery of neural function in an animal model of SCI (Shi et al., 1997). Fehlings and Nashmi (1995), in another in-vitro experiment, have shown that 4-aminopyridine can partly restore electrophysiological function in the injured spinal cord. In addition to pharmaceutical interventions, the recruitment of myelin-producing cells is another strategy for enhancing axonal remyelination following SCI. Utzschneider and colleagues (1994) transplanted glial cells into the spinal cords of neonatal rats with myelin deficiency, and demonstrated enhanced myelin formation and increased conduction velocity in surviving axons. Taken together, these findings provide considerable evidence supporting the notion that improving axonal myelination is of major importance for the recovery of neural function in SCI.
Mechanisms of Axonal Demyelination
The myelin-forming cells of the CNS come exclusively from the oligodendrocyte population. These myelinating cells project extensions of their cytoplasmic membrane that wrap segments of nearby axons to form the myelin sheath (Morell et al., 1994; Peters et al., 1991; Abe et al., 1999). It is well known that injuries secondary to the initial insult in SCI exacerbate the impairment of neural function, and oligodendrocytes in the area of the lesion appear to be highly vulnerable to such injury. Consequently, axonal demyelination is probably related to the demise of oligodendrocytes in SCI. A series of studies, discussed below, have presented convincing evidence in support of this idea.
Some groups have documented that injury to the dorsal column brings about the degeneration of white matter and the death of intrafascicular oligodendrocytes (Crowe et al., 1997), and others have found that oligodendrocyte apoptosis can be detected as early as the first 24 h after injury and persists for at least 3 weeks (Beattie et al., 2000). Warden and co-workers (2001), in a study of the relationship of apoptosis to the wallerian degeneration of white matter, observed substantial cellular apoptosis in areas of wallerian degeneration within tracts of white matter in SCI, and considered oligodendrocytes to be responsible for a portion of the cellular apoptosis seen in these areas. Additionally, a clinical study found apoptosis of oligodendrocytes in human SCI. The apoptotic cells were seen on the borders of the epicenters of spinal lesions and in the vicinity of white matter, and appeared likely to have come from the oligodendrocyte population (Emery et al., 1998).
Continuing progress in the neurosciences has gradually revealed the mechanisms underlying the death of oligodendrocytes in SCI. Excessive concentrations of excitatory amino acids, insufficiency of trophic factors, and activation of caspases, have been proven to play important roles in the apoptosis of oligodendrocytes following SCI. Xu and colleagues (2004) showed that SCI in rats releases sufficient glutamate to kill oligodendrocytes and damage white matter in the spinal cord. This work clearly demonstrates that following SCI, the accumulation of excitatory amino acids at the lesion site reaches concentrations toxic to oligodendrocytes. Additionally, Shuman and associates (1997) showed that apoptosis of oligodendrocytes after SCI results in part from an insufficiency of trophic support, and proposed that the loss of oligodendrocytes is associated with the demyelination of surviving axons.
Caspases are well known to play vital roles in cellular apoptosis, and in this context the activation of caspases has been shown to be involved in the apoptosis of oligodendrocytes in SCI. In in-vivo studies with a rat model of contusive SCI, immunohistochemical analysis has indicated that caspases are primarily activated in both neurons and oligodendrocytes, and that the administration of caspase inhibitors immediately after the initial injury can improve locomotor function following SCI (Knoblach et al., 2005; McEwen and Springer, 2005). Casha and associates (2001) found that both Fas and p75 protein are activated in apoptotic oligodendrocytes in a rat model of SCI, and that downstream caspases 3 and 8 are active at the time of maximal apoptosis.
Collectively, the findings described above support the conclusion that the demyelination of axons in SCI is closely related to the dysfunction of oligodendrocytes, and that the apoptosis of oligodendrocytes is an important pathologic factor in such axonal demyelination.
Improving Axonal Myelination
The loss of oligodendrocytes and subsequent demyelination of axons in SCI remain an insidious and challenging problem in neurological research. Fortunately, recent advances in neuroscience have brought hope into this once daunting field. The acute phase of SCI seems to be followed by a therapeutic window of opportunity for minimizing the harmful effects of secondary injury to the spinal cord. Moreover, clinical autopsy studies have confirmed the preservation of unmyelinated axons in the lesion area in SCI, which could serve as a therapeutic target for the improvement of neural function. At present, the approaches to improving axonal myelination following SCI can generally be classified into two therapeutic strategies: preventing axonal demyelination, and enhancing axonal remyelination.
Preventing Axonal Demyelination
The death of oligodendrocytes and consequent demyelination following SCI severely impair neural function. Theoretically, protecting myelinating cells from secondary injury and enhancing their survival are likely to decrease axonal demyelination and consequently facilitate functional recovery. The presence of as few as 5–10% of myelinated axons at the lesion site in SCI can preserve some neural function. Thus saving the mature oligodendrocytes that wrap viable axons at the lesion site has therapeutic potential in SCI. To date, several agents have proved to have neuroprotective effects on oligodendrocytes and to prevent axonal demyelination following SCI.
A lack of neurotrophic factors at the lesion site is partly responsible for the death of oligodendrocytes in SCI. The provision of neurotrophic substances after SCI can rescue some dying oligodendrocytes. In an early study of this, it was reported that continuous intrathecal infusion of brain-derived neurotrophic factor immediately after SCI in rats significantly reduced the number of apoptotic oligodendrocytes in white matter of the cord (Koda et al., 2002). This finding indicates that supplementation with neurotrophic factors can suppress the delayed apoptosis of oligodendrocytes in SCI. Additionally, Jiang and associates (2003) reported that the administration of guanosine for 7 consecutive days after SCI in a rat model improved locomotor function and axonal remyelination, and demonstrated that this myelinogenesis was associated with an increase in the number of oligodendrocytes in the treated groups of animals. Taken collectively, these research findings suggest a key role for the administration of neurotrophic factors in treating SCI.
Minocycline, a second-generation tetracycline, has been proven to be neuroprotective in SCI. Some groups have investigated whether minocycline protects oligodendrocytes and prevents axonal demyelination in SCI. Stirling and colleagues (2004) showed that injection of minocycline after transection of the dorsal column can reduce the numbers of apoptotic oligodendrocytes in segments of nerve fiber tracts both proximal and distal to the site of SCI in rats, and found that footprint analysis demonstrated functional recovery in minocycline-treated animals. Several other groups of investigators have conducted experiments to further explore the mechanisms underlying the neuroprotective effects of minocycline on oligodendrocytes in SCI. Teng and co-workers (2004) showed that minocycline exerts its effects in SCI by inhibiting the release of mitochondrial cytochrome c. Delivery of minocycline significantly reduced the cytosolic concentration of cytochrome c at the epicenter of the lesion site in SCI in rats, and markedly improved neural function and reduced late-phase tissue loss, with significant sparing of white matter and motoneurons. More recently, Yune and associates (2007) demonstrated that minocycline contributed to functional recovery after SCI in rats by reducing oligodendrocyte apoptosis through the inhibition of pro-nerve growth factor production in microglia, which plays a known role in oligodendrocyte apoptosis following SCI.
Leukemia-inhibitory factor (LIF), a pleiotropic cytokine, is a strong candidate substance for supporting oligodendrocyte survival in SCI. LIF potentiates the differentiation and survival of oligodendrocyte precursors, and also prevents oligodendrocyte apoptosis in response to growth factor removal as well as cytotoxic challenge (Vos et al., 1996; Kerr and Patterson, 2005; Slaets et al., 2008). LIF works partly by inducing the JAK/STAT and Akt signaling pathways, as well as by potentiating expression of the anti-apoptotic molecule cIAP2, and its systemic delivery has been found to reduce oligodendroglial death and prevent oligodendrocyte apoptosis in animal models of SCI. LIF-mediated reduction of oligodendrocyte apoptosis substantially decreases axonal demyelination, as indicated by the preservation of lamellated myelin surrounding viable axons in the spinal cords of rats with SCI (Kerr and Patterson, 2005; Azari et al., 2006).
Because the activation of caspases is a major contributor to oligodendrocyte apoptosis among the injuries secondary to SCI, it is likely that oligodendrocytes can be preserved by inhibiting the activation of caspases (Dong et al., 2003). The immunosuppressant drug FK506 has been used to inhibit the activation of caspases in oligodendrocytes following SCI. This was found to spare numerous oligodendrocytes in regions proximal and distal to the epicenter of the injury site in treated groups of animals. These results suggest that treatment with FK506 may be a way of reducing the numbers of oligodendrocytes expressing activated caspase-3, thus increasing the numbers of surviving oligodendrocytes in the cord white matter after SCI (Nottingham et al., 2002). In other research, the impairment of motor function and histopathologic changes following contusive thoracic SCI were reduced in a transgenic mouse model expressing p35, a broad-spectrum caspase inhibitor (Tamura et al., 2005; Hisahara et al., 2000). A larger number of oligodendrocytes and a lesser extent of demyelination were simultaneously observed in the transgenic mice as compared to controls (Tamura et al., 2005). The inhibition of oligodendroglial apoptosis can therefore also reduce the demyelination of axons and consequently the magnitude of functional impairment in SCI.
Cell Transplantation for Remyelination
Beyond the apoptosis of oligodendrocytes, the loss of myelin-forming cells and subsequent demyelination of surviving axons further impairs neural function in SCI. Recent studies point to cell replacement as one of the most promising strategies for facilitating axonal remyelination and restoring conductive function following such injury (Table 1). In order to remyelinate anatomically preserved but physiologically disrupted axons, novel therapeutic interventions have employed the transplantation of myelinating cells. At present, the cells used for such transplantation come mainly from populations of Schwann cells, olfactory ensheathing cells, oligodendrocyte precursors, and embryonic or neural stem cells (Kocsis et al., 2002).
Schwann cells are the myelin-forming cells in the peripheral nervous system, and they play a vital role in the remyelination of demyelinated axons after peripheral nerve injury. In this regard, Schwann cells are considered attractive cellular candidates for remyelination therapy following injury to the CNS. However, their unfavorable integration into the CNS, resulting from inhibition by astrocytes, greatly limits their utility for grafting to treat injury to the CNS. Currently however, Schwann cells can be genetically modified through advanced techniques to improve their adaptability for cell transplantation into the CNS. These genetically engineered Schwann cells, expressing on their surfaces the polysialylated form of neural cell adhesion molecule, have been transplanted in a mouse model of SCI, in which they were found to promote significantly more rapid functional recovery than that seen in controls. Moreover, the morphologic investigation indicated that the improvement of locomotor function that followed transplantation of these Schwann cells correlated with the enhanced axonal remyelination produced by the engrafted cells (Papastefanaki et al., 2007).
The transplantation of olfactory ensheathing cells (OECs) has emerged as another promising strategy for promoting recovery after SCI. OECs seem to be better than Schwann cells as candidates for producing extensive remyelination and axonal regeneration. In animal models of SCI, implanted OECs can form myelin sheaths around demyelinated axons and migrate extensively throughout the length of the spinal cord, and preliminary clinical trials suggest a therapeutic role for transplantation of these cells in human SCI (Bartolomei and Greer, 2000; Santos Benito and Ramon Cueto, 2003; Barnett and Riddell, 2007). In an in-vivo study in which OECs expressing green fluorescent protein (GFP) were transplanted into the transected spinal cords of rats, the implanted cells survived in the area of the cord lesions and distributed longitudinally along axons bridging the transection site (Sasaki et al., 2004). Locomotor function analysis showed significant improvement in the treated groups of animals. Myelinated axons spanning the lesion were seen in discrete bundles encapsulated by a cellular element, and microscopic study with anti-GFP antibody indicated that a majority of the peripheral-like (i.e., large cytoplasmic and nuclear regions surrounding the myelinated axons) myelinated axons were derived from the donor OECs (Sasaki et al., 2004).
Neural precursor cells (NPCs) are multipotent stem cells with the ability to differentiate into all three major types of neural cells. Recently, (Karimi-Abdolrezaee and colleagues 2006) transplanted adult brain-derived NPCs isolated from yellow fluorescent protein–expressing transgenic mice into the injured spinal cords of adult rats. A substantial number of surviving NPCs were found in the injured cords up to 10 weeks after transplantation, and the cells had also migrated both rostrally and caudally. The NPCs had principally integrated along white matter tracts and showed close contact with host axons. Approximately 50% of the grafted cells appeared as either oligodendroglial precursor cells, or as mature oligodendrocytes that expressed myelin basic protein (MBP) and ensheathed axons, with a concurrent improvement in neural function in the treated rats. These findings provide strong evidence for the feasibility and practicality of transplantation of NPCs as a means of cell-based remyelination in SCI.
As discussed earlier, the loss of oligodendrocytes leads directly to axonal demyelination in SCI, and the death of oligodendrocytes and subsequent demyelination of axons contribute significantly to the neurological impairment seen in SCI. Accordingly, replenishment of lost oligodendrocytes and remyelination of surviving axons can be expected to bring significant improvements in functional outcome following SCI. In a study intended to assess the potential of cells of oligodendroglial lineage for restoring axonal myelination in SCI, (Rosenbluth and associates 1997) implanted glia derived from transgenic mice into the lesion sites of rats with SCI. After transplantation, the grafted cells were identified in cord segments rostral and caudal to the implantation site, with most of the cells being found within the white matter defects in the cord. Furthermore, some implanted cells were oriented parallel to the fiber tracts and had projected processes similar to myelin segments. Thus glial transplantation is a feasible means of replacing the myelin-forming cells lost in SCI with donor cells of oligodendrocyte lineage that are capable of forming myelin and potentially improving neural function in SCI (Rosenbluth et al., 1997).
In another study, (Bambakidis and Miller 2004) transplanted oligodendrocyte precursors into the lesion sites of rats with contusive injuries of the spinal cord. The treated rats showed significant improvements in locomotor function, and their electrophysiological outcomes were comparable to those of control animals by 28 days after transplantation. Immunohistochemical analysis demonstrated survival, proliferation, and migration of the implanted cells at the transplant site, and histologic examination found significantly more preserved white matter in animals that showed improvements in neural function (Bambakidis and Miller, 2004). Human embryonic stem-cell-derived oligodendrocyte progenitor cells (OPCs) have been studied for potential use in recruiting myelin-forming cells in SCI. The transplantation of OPCs into adult rats with SCI enhanced axonal remyelination and promoted improvements in neural function (Keirstead et al., 2005). The implanted cells survived, redistributed over short distances, and differentiated into mature oligodendrocytes. The treated animals exhibited enhanced remyelination and substantially improved locomotor function (Keirstead et al., 2005). Oligodendrocyte-type 2 astrocyte (O-2A) progenitor cells have also been investigated for transplantation in SCI (Lee et al., 2005). O-2A cells implanted into rats with SCI were detected mainly at the injury epicenter and nearby, and double immunostaining indicated that the transplanted cells differentiated into mature oligodendrocytes. Improvements in locomotor and electrophysiologic function were seen in the O-2A-cell recipient groups (Lee et al., 2005). Collectively, these findings indicate that transplanted oligodendrocyte precursors can remyelinate demyelinated axons and improve the neural function of the injured spinal cord.
Pluripotent cells other than neural cells have also been investigated for transplantation in treating the axon demyelination in SCI. For example, the transplantation of human umbilical cord blood stem cells (hUCBs) has shown therapeutic promise for axonal remyelination in SCI (Dasari et al., 2007). Implanted hUCBs were found to primarily differentiate into oligodendrocytes, and to enhance the synthesis of MBP and the proteolipid protein of myelin in cord-injured areas of treated rats. Ultrastructural analysis further showed that hUCBs formed myelin sheaths of morphologically normal appearance around axons in the lesion area, and significantly improved locomotor function in the treated rats (Dasari et al., 2007). Bone marrow contains a population of pluripotent cells that can differentiate into a variety of cell types, including neural cells. The systemic delivery of bone marrow cells has been found to improve the neural function of rats with contusive SCI. Remyelination of demyelinated areas of spinal cord was observed throughout the focal lesions of SCI after delivery of these cells, and the conduction velocity in remyelinated axons was improved (Akiyama et al., 2002).
In sum, this review details compelling evidence that there is great therapeutic potential for transplanting myelin-forming cells to remyelinate surviving axons and restore neural function after SCI.
Conclusion
Preserving oligodendrocytes and the myelin sheath can be expected to bring significant improvement of neural function in SCI. A therapeutic window of opportunity for reducing the death of oligodendrocytes and counteracting the demyelination of surviving axons appears to exist after the acute phase of injury, with subsequent benefit to the recovery of neural function in SCI. Because spontaneous remyelination in SCI is severely limited, cell transplantation should be considered an attractive approach for oligodendrocyte replacement and recruitment, and for enhancing axonal remyelination in SCI. In the future, replenishing oligodendrocytes and restoring axonal myelination may take its place among the strategies for treating SCI. The therapeutic potential of cell-transplantation strategies in SCI will probably depend on the success with which they can be combined with other treatment strategies to achieve significant remyelination of surviving axons, as well as axon regeneration in SCI, and to re-establish functional connections across the area of injury.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.