
Abstract
Restraint stress (RS) protects auditory function against acoustic trauma by activating glucocorticoid receptors (GR) in the cochlea. In a search for the signaling pathways downstream to GR that may be involved in RS-induced protection we report here (1) a downregulation of phosphorylated extracellular signal-regulated kinases 1 and 2 (pERK 1/2) after the combined treatment of RS and acoustic trauma; (2) activation of phospho-p38 in the auditory nerve after RS; (3) the abolition of these two effects by pretreatment with metyrapone (an inhibitor of corticosterone synthesis) and RU486 (a GR antagonist); and (4) no activation of c-jun-N-terminal kinases 1 and 2 (JNK 1/2), ERK, or p38 after acoustic trauma alone. Thus we demonstrate a GR-dependent ERK-mediated pathway that modulates auditory function after RS and acoustic trauma. These findings reveal new mechanisms that underlie hearing loss and will have implications for the development of pharmacological strategies for protecting against acoustic trauma.
Introduction
R
One important signaling pathway that is involved in the response to stressful stimuli is the mitogen-activated protein kinase (MAP kinase) system (Kyriakis and Avruch, 2001). The activation of different MAP kinases can result in cell growth, differentiation, or death, a response that depends on the particular intracellular pathway that becomes activated, the intensity of the stimulus, and the cell type. It has also been found that stress increases the expression of c-jun-N-terminal kinase (JNK) in the brain (Nankova et al., 1998), and modulates the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK 1/2) (Meller et al., 2003). MAP kinases are expressed in auditory brainstem neurons (Suneja and Potashner, 2003; Suneja et al., 2005), as well as in the cochlea (Hess et al., 2002). Protection against hearing loss induced by ototoxic antibiotics has been found after pretreatment with JNK inhibitors (Pirvola et al., 2000). Thus GR and MAP kinases have independently been demonstrated to protect against hearing loss induced by acoustic trauma. The goal of the present study was to determine if MAP kinases are involved in protecting against hearing damage in a GR-dependent manner.
Methods
Animals
A total of 106 CBA male mice (B&K Universal AB, Sollentuna, Sweden) aged 10–12 weeks (20–25 g) without any evidence of middle ear infection were used in this study.
The animals were housed in groups of five animals per cage on an artificial light/dark cycle (12/12 h, lights on at 07:00 h), with free access to food and water. The Northern Stockholm Ethical Committee approved the care and use of animals for this experiment.
Pharmacological manipulations
The glucocorticoid synthesis inhibitor metyrapone (Met) (2-methyl-1,2-di-3-pyridyl-1-propanone; Sigma, St. Louis, MO), and the glucocorticoid receptor antagonist RU486 (Roussel Uclaf, Romain Ville, France) were used as previously described (Tahera et al., 2006a). Met (200 mg/kg in water solution) and RU486 (100 mg/kg in vegetable oil) were injected intraperitoneally (IP) and subcutaneously (SC), respectively.
Experimental procedure
The animals were divided in two groups: one received water and oil injections, and the other received Met and RU486 solution injections 1 h 30 min prior to all manipulations. Figure 1 illustrates the different treatments used in this study. The control animals were placed in single cages for sham restraint and an acoustic chamber without sound for sham trauma. For RS, the animals were placed in 50-mL plastic tubes with ventilation holes and placed in a soundproof booth for 4 h. The animals did not have access to food or water during these 4 h. After the cessation of restraint they were placed in an acoustic chamber to mimic the conditions of the trauma group, but no sound was generated. For the acoustic trauma, the animals were placed in single cages with no food or water for 4 h to mimic conditions of RS (sham RS). Acoustic trauma (45 min) as described below was used immediately after sham restraint. The RS + trauma animals were subjected to RS as described, and immediately after cessation of the RS they were exposed to acoustic trauma. The animals were anesthetized immediately after cessation of these procedures and sacrificed within approximately 15 min.

Schematic diagram showing the experimental procedure. Each group was divided into two subgroups, treated either with vehicle (saline + mineral oil) or Met + RU486. The animals were placed in their home cages after injection, and 1.5 h thereafter were subjected to restraint stress (RS) or sham RS (placed in a single cage without access to food or water for 4 h in a soundproof chamber). Then they were moved into a mesh cage for exposure to acoustic trauma (100 dB SPL, 6–12 kHz, 45 min) or without trauma (Sham). All animals were anesthetized and killed immediately after the cessation of acoustic or sham trauma.
Acoustic trauma
The acoustic trauma used in this study was performed as previously described (Tahera et al., 2006a). Briefly, free field broadband noise at 6–12 kHz was used for 45 min at an intensity of 100 dB sound pressure level (SPL). The stimulus was generated by a noise generator (Hewlett Packard 33120 A; Hewlett-Packard, USA), and delivered from the upper corners of the chamber by four speakers. Calibration of the sound-exposure levels was performed with a 12.5-mm condenser microphone (model 2213; Bruel and Kjaer, Denmark). The acoustic trauma used in this study caused a temporary change in auditory thresholds. At the end of the exposure the animals were anesthetized with ketamine (50 mg/kg) and xylazine (10 mg/kg) injected IP, and sacrificed by transcardiac perfusion with ice-cold buffered PBS with heparin immediately after trauma or sham trauma.
Tissue collection and protein isolation
The cochleae were immediately removed after PBS perfusion, placed in an ice-cold solution containing complete protease inhibitor (Roche Diagnostics, Mannheim, Germany), 1 mM DTT, and 1 mM Na3VO4. The cochleae were perfused through the round window and the bony shell was removed. The cochleae were divided into two fractions: the auditory nerve (the modiolus with the spiral ganglion) and the cochlear tissues (the lateral wall and the organ of Corti). The collected tissues were preserved at −70°C. To obtain a total protein extract, the cochleae of three mice were pooled. The tissue was lysed for 40 min on ice in lysis buffer containing: 20 mM Tris-HCl (pH 7.0), 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 2.5 mM sodium pyrophosphate, 1 mM sodium glycerophosphate, 1 mM DTT, 1 mM Na3VO4, and 1% v/v Triton-X-100 and complete protease inhibitor (Roche Diagnostics). The mixture was centrifuged at 15,000 × g for 5 min at 4°C to yield lysates. All lysates were frozen and stored at −80°C until assayed. Protein concentration was determined according to the Bradford method, using bovine serum albumin as a standard.
The cytosolic and nuclear fractions were obtained as previously described (Meltser et al., 2008). Briefly, the cochleae were pooled and placed in lysis buffer containing 25 mM Tris pH 7.5, 50 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, and complete protease inhibitor. The tissue was homogenized on ice, centrifuged at 1700 × g for 10 min at 4°C, and supernatant was collected for the cytosolic fraction. The precipitate was re-suspended in a new buffer (25 mM Tris [pH 7.5], 0.42 M NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 25% sucrose, 1 mM DTT, and complete protease inhibitor), lysed for 40 min on ice, centrifuged at 20,000 × g for 30 min at 4°C, and the supernatant was collected as the nuclear fraction. Both cytosolic and nuclear fractions were concentrated using the PAGE Prep Advance Kit (#89888; Pierce, Rockford, IL). The concentration of protein was measured using the BCA™ Protein Assay Kit (#23227, Pierce).
Western blot analysis
Concentrated nuclear or cytosolic protein fractions containing 15 μg total protein per sample were resuspended in NuPAGE® LDS Sample Buffer (catalogue # NP0007; Invitrogen, Carlsbad, CA) supplemented with DTT, heated for 5 min at 95°C, and separated by SDS-PAGE electrophoresis with 4% spacer and 12% (w/v) separating polyacrylamide gel. The protein extract of 10 μg of total protein was loaded on each lane. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Amersham Pharmacia Biotech, Little Chalfont, U.K.) by semi-dry electroblotting. For detection, the PVDF membranes were blocked for 2 h in TBST (20 mM Tris and 150 mM NaCl [pH 7.6]), containing 0.1% (v/v) Tween 20 supplemented with 5% nonfat dry milk prior to overnight incubation with primary antibodies. After three 10-min washing steps using TBST, the PVDF membranes were incubated for 1 h with peroxidase-conjugated anti-rabbit or anti-mouse IgG goat antibodies (Pierce). Thereafter, the PVDF membranes were washed three times for 10 min and two times for 1 min with TBST or TBS, respectively, to remove residual antibody. The PVDF membranes were developed with an enhanced chemiluminescence Western blot detection kit (Pierce SuperSignal®) and exposed to Lumi-Film chemiluminescent detection film (Roche Diagnostics). The protein loading was verified by staining with MemCode (Pierce). The antibodies used for Western blot were as follows (suppliers, catalogue numbers, and dilutions are given in parentheses): anti-ERK (Santa Cruz Biotechnology, Santa Cruz, CA; sc-154; 1:800), anti-pERK (Santa Cruz Biotechnology; sc-7383; 1:200), anti-JNK (Santa Cruz Biotechnology; sc-571; 1:200), anti-pJNK (Santa Cruz Biotechnology; sc-6254; 1:200); anti-GR (Affinity BioReagents, Golden, CO; PA1-511A, 2.5 μg/mL), anti-pp38 (Cell Signaling Inc., Beverly, MA; #9211, 1:1000), anti-TATA binding protein (TBP) (Abcam Ltd., Cambridge, UK; #ab818, 1:1000), and anti-GAPDH (Abcam Ltd.; #ab9484, 1:1000). Quantification of Western blots was performed using Tina software (Raytest, Isotopen Messergäte GmbH, Munich, Germany).
Immunocytochemistry
Immunocytochemical analysis was performed on the cochleae from the vehicle-treated group. After fixation (1 h) the cochleae were decalcified using Rapid Decalcifying Agent (Apex Engineering Products Corp., Plainfield, IL), and then placed in 10% sucrose for 12–24 h at 4°C, followed by 20% sucrose for 24 h. The specimens were flash frozen and stored at −70°C. Serial mid-modiolar sections (14-μm thickness) were cut on a cryostat (HM 500M; Zeiss, Germany) at −24°C. The Vectastain® ABC standard kit (Vector Laboratories, Inc., Burlingame, CA) was used for rabbit primary antibody as described previously (Tahera et al., 2006a), and the Vector® M.O.M Immunodetection Kit (Vector Laboratories) was used for mouse primary antibody by following the manufacturer's instructions. The last step was performed with DAB nickel and afterward was counterstained with cresyl violet for pJNK and synaptophysin. For staining with anti-pERK and anti-pJNK the same antibodies were used as for Western blot in the same concentrations, and anti-GR was applied in a concentration of 5 μg/mL. Anti-synaptophysin antibody was supplied by Sigma (#S5768) and used in a dilution of 1:200, and anti-phospho-p38 was supplied by Cell Signaling (#4631) in a dilution of 1:50.
Statistical analysis
Data are presented as mean values ± SD. The comparisons of the means were performed with two-way ANOVA with Tukey correction. A value of p < 0.05 was considered statistically significant. SigmaStat version 2.03 (Systat Inc., Richmond, CA) was used for statistical analysis.
Results
GR localization and activation in the cochlea
Expression of GR was found in both neuronal and non-neuronal cochlear tissues. In the vehicle-treated group, immunostaining was found in the nucleus of inner and outer hair cells, supporting cells (Fig. 2A), and spiral ganglion neurons (Fig. 2B). Positive staining was also found in fibrocytes of the spiral ligament (Fig. 2C). The specificity of anti-GR antibody used for the staining was verified by Western blot (Fig. 2D).
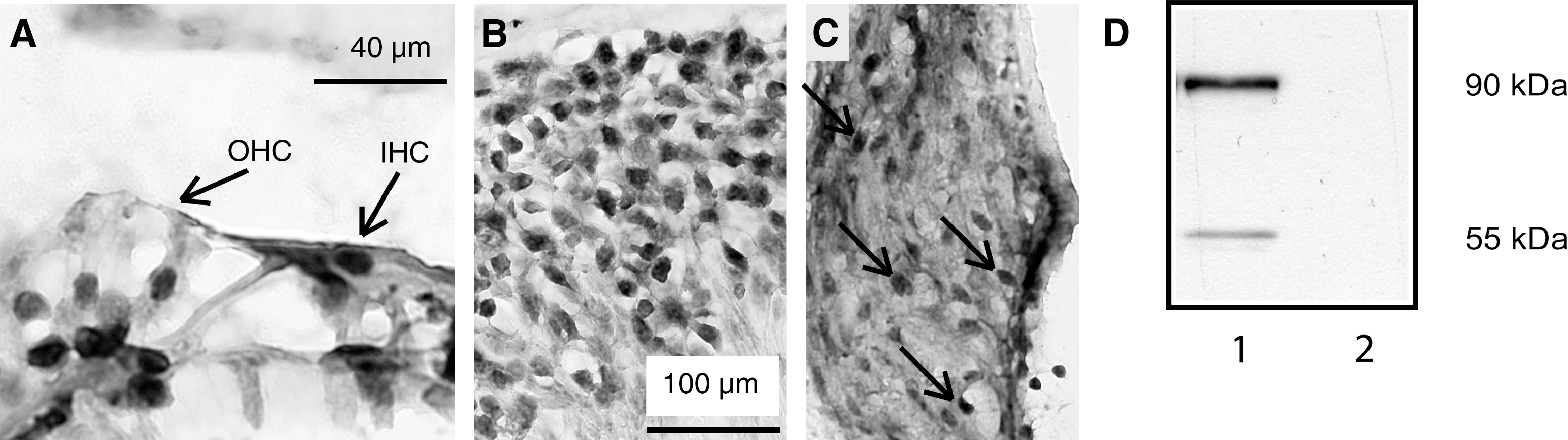
Immunocytochemical localization of the glucocorticoid receptor (GR) in mouse cochleae. (
In order to determine the effect of acoustic trauma on GR nuclear translocation, Western blot analysis was performed on nuclear and cytosolic fractions of cochlear homogenates. The expression of TBP and GAPDH served as quality controls for the nuclear and cytosolic fractions, respectively (Fig. 3B). In vehicle-treated animals, the GR expression in the nuclear fraction was lower than in the cytosolic fraction (Fig. 3A, lane 1). Met + RU486 treatment did not change the GR expression in either fraction (lane 2) compared to the vehicle group. Vehicle + acoustic trauma (lane 3) and vehicle + RS (lane 4) resulted in an increase in GR in the nuclear fraction, and a decrease in the cytosolic fraction, compared to the vehicle-alone group. Met + RU486 treatment combined with acoustic trauma (lane 5), or Met + RU486 treatment with RS (lane 6) did not change the GR in the nuclear or cytosolic fraction compared to the vehicle-alone groups. These results demonstrate that RS and acoustic trauma increase GR expression in the nuclear fraction, and decrease GR in the cytosolic fraction, and that Met + RU486 treatment blocked GR translocation into the nucleus after RS or acoustic trauma.

(
Total ERK and pERK expression in the cochlea
Immunocytochemical analysis showed a moderate expression of pERK in the nuclei of the inner and outer hair cells, the supporting (Deiters') cells (Fig. 4A), and spiral ganglion neurons (Fig. 4B). A few fibrocytes of the spiral ligament also contained pERK, while the stria vascularis was negative (Fig. 4C). Total ERK expression in the nerve and cochlear fractions was not altered after any treatment according to Western blot analysis, therefore the total ERK levels were used as a loading control for phospho-ERK (Fig. 4E). RS alone or acoustic trauma alone did not change the expression of pERK in the nerve fraction, while the combined treatment of RS and trauma significantly decreased pERK compared to vehicle alone (p < 0.0001) and RS (p < 0.0001) (Fig. 4D). In the presence of Met + RU486, the control group showed a significant decrease in pERK expression compared to vehicle alone (p < 0.001). Met + RU486 treatment prior to RS also reduced pERK levels compared to vehicle + RS. pERK expression after Met + RU486 + RS + trauma was significantly higher than after Met + RU486 alone (p < 0.001), Met + RU486 + RS (p < 0.001), and vehicle + RS + trauma (p < 0.0001).

(
The expression of pERK in cochlear tissues had similar patterns of expression to those found in the nerve. RS alone did not change the levels of pERK, while RS + trauma significantly decreased it compared to vehicle (p < 0.001), or vehicle + RS (p < 0.001; Fig. 4D). The significant decrease in pERK after the combination of RS and trauma reflects the complexity of the regulation of ERK phosphorylation in the peripheral auditory nerve and cochlear tissues. Met + RU486 treatment alone, or when given prior to RS, significantly decreased pERK in cochlear tissues compared to vehicle alone or vehicle + RS (p < 0.001). Met + RU486 treatment prior to acoustic trauma resulted in an upregulation of pERK compared to Met + RU486 alone (p < 0.001). Met + RU486 treatment prior to RS + trauma resulted in a significant upregulation of pERK compared to vehicle + RS + trauma (p < 0.001).
These findings suggest that the suppression of GR activity results in the suppression of pERK in the absence of acoustic trauma. When acoustic trauma is applied in the presence of a GR-antagonist, an elevation in pERK occurs. In the absence of Met + RU486, trauma had no effect on pERK, suggesting that activated GR prevents pERK elevation. The combination of RS and trauma in the Met + RU486 group resulted in the same effect as trauma alone, and reversed the pERK reaction compared to vehicle-pretreated animals.
Phospho-p38 expression in the cochlea
The expression of phospho-p38 (pp38) was localized to the organ of Corti (Fig. 5A) and spiral ganglion neurons (Fig. 5B), while the spiral ligament and stria vascularis were negative (not shown). Western blot analysis revealed an upregulation of the expression of pp38 in the auditory nerve after RS compared to the vehicle group (p = 0.014), and compared to trauma (p = 0.009; Fig. 5C and D). Acoustic trauma alone did not change pp38 expression compared to vehicle. Met +RU486 treatment alone or in combination with either RS or acoustic trauma had no significant effect on pp38 expression compared to vehicle alone. Thus RS induced an upregulation of pp38 in the peripheral auditory nerve, and this induction was abolished by Met + RU486. In the auditory nerve, combined treatment with RS + trauma resulted in a decrease in pp38 (p = 0.008), which was reversed by Met + RU486 treatment. No change in the expression of pp38 in the cochlear tissues was found in any of the vehicle-treated groups (Fig. 5C). The effect of Met + RU486 treatment alone did not differ from that seen in the vehicle group. A significant difference between the vehicle + RS and the Met + RU486-treated RS group was found (p < 0.001). Met + RU486 + RS + trauma caused an upregulation of pp 38 compared to Met + RU486 +RS (p = 0.004). These data suggest that RS, but not acoustic trauma, increased pp38 expression in the auditory nerve in a GR-dependent manner.

(
Total JNK and pJNK expression in the cochlea
The immunocytochemical analysis revealed active JNK (pJNK) in the organ of Corti (Fig. 6A). Staining was localized to the base of the outer hair cells. Since it was difficult to discriminate if the staining was in the nerve endings or the supporting (Deiters') cells, neighboring sections were labeled for synaptophysin and pJNK. We found synaptophysin staining in the nerve endings below the outer and inner hair cells (Fig. 6B). Pseudocolor merged images of pJNK (green) and synaptophysin (red) indicated that there was pJNK labeling was in the supporting Deiters' cells (Fig. 6C). The inner hair cells and their supporting cells were negative for pJNK, as was the spiral ligament, stria vascularis, and spiral ganglion (not shown). Western blot demonstrated the total JNK expression in the whole-cell protein extract from the nerve (Fig. 6D, two left panels) and cochlear tissues (Fig. 6D, two right panels). The levels of pJNK after the different treatments were low, and none of the treatments resulted in a detectable change.

(
Discussion
pERK is downregulated after RS and trauma in a GR-related manner
RS is known to protect the cochlea against acoustic trauma–induced hearing loss (Tahera et al., 2006c; Wang et al., 2002). Here we show that the protective effect of RS on the auditory system involves a GR-dependent ERK signaling pathway (Fig. 7). Table 1 is a summary of the results obtained for all groups compared to vehicle alone. We demonstrated a downregulation of pERK in the cochlea after the combined treatment of RS and acoustic trauma, an effect that was abolished after Met + RU486 treatment. We also demonstrated that the expression of pERK in the cochlea was detected only after vehicle injection. This is in contrast to other studies that could not demonstrate any expression of pERK in the intact cochlea. One reason for this difference could be that in the present study, the mice were injected with vehicle, which elicits a stress response, while other studies analyzed untreated cochlea (Hess et al., 2000). No increase in pERK after acoustic trauma was found, suggesting that the trauma used in our study had no immediate effect on ERK activation. Another explanation is that the activation of ERK may occur within the first seconds after trauma very briefly, and we were unable to detect it due to the 15-min period between the cessation of trauma and cochlear extraction. It is also important to mention that the acoustic trauma used in our study leads to reversible metabolic changes in cochlear tissues and temporary hearing loss, with complete recovery of hearing by 48 h (Tahera et al., 2006a).
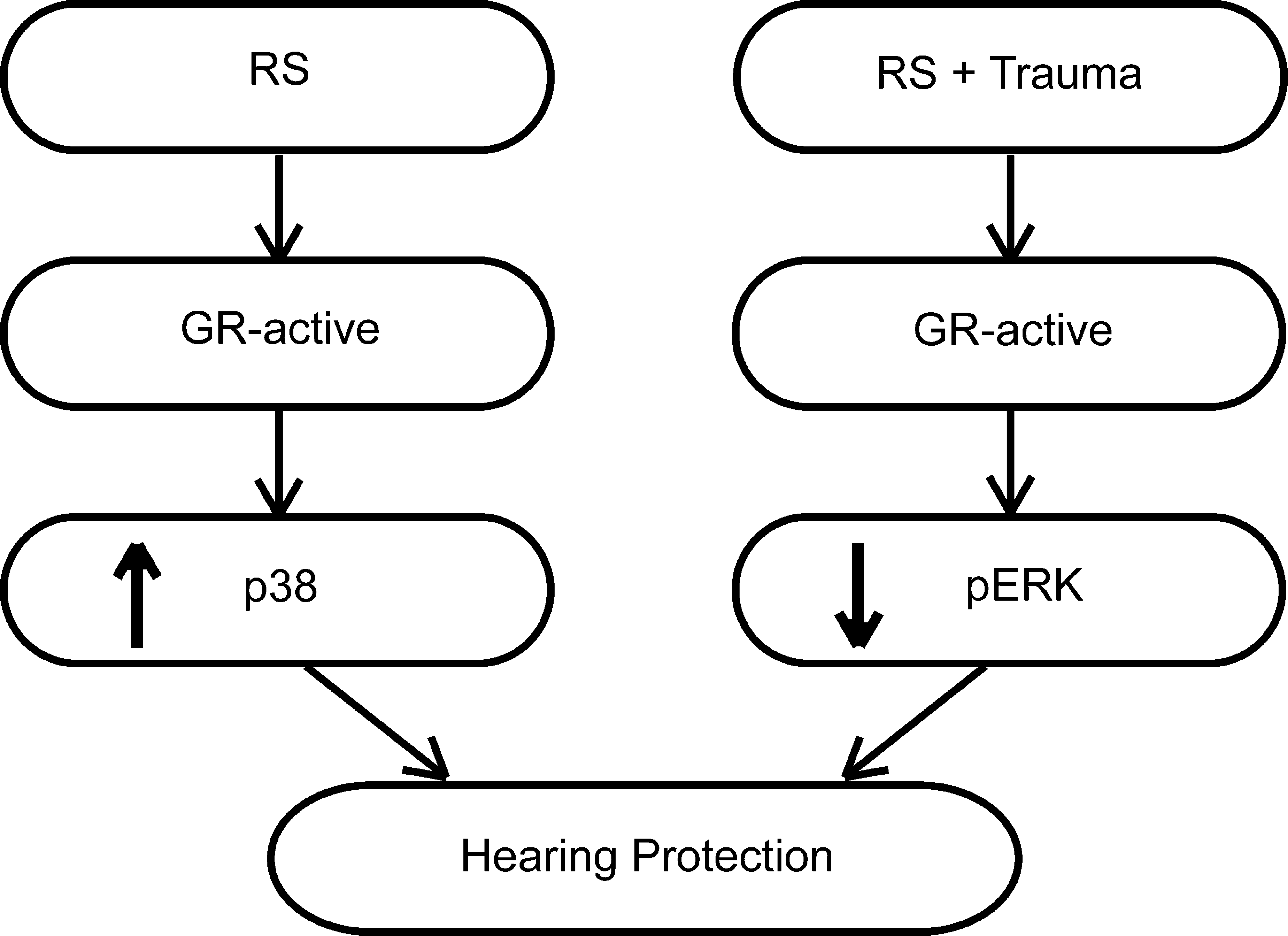
Summary of the results, showing a hypothetical pathway for the cross-talk between the glucocorticoid receptors (GR) and MAP kinases. Restraint stress (RS) alone (left) activates GR, leading to p38 phosphorylation (activation) and protection against acoustic trauma. When GR are blocked by Met + RU486 there is no activation of GR or p38, and hearing loss is exacerbated. When RS is followed by acoustic trauma GR are activated, resulting in a decrease in pERK and protection against acoustic trauma. When GR are inhibited, there is an increase in pERK and hearing loss is exacerbated.
No change compared to vehicle alone.
It is well established that ERK is involved in the cell death of neurons (Ho et al., 2008; Satoh et al., 2000), glutamate-induced exitotoxicity (Lubin et al., 2005; Vanhoutte et al., 1999), and the production of proinflammatory cytokines (Jeong et al., 2005; Nam, 2006). The activation of ERK in the cochlea has been found after hypoxia and mechanical damage (Hess et al., 2002; Lahne and Gale, 2008). On the contrary, it can also be activated by growth factors in neurons, which in turn inhibits apoptosis and promotes neuronal outgrowth (Aletsee et al., 2002; Arthur et al., 2006; Hetman et al., 1999; Lallemend et al., 2003; Xia et al., 1995). It has also been shown that ERK is necessary for cochlea development (Urness et al., 2008). Here we showed that the active forms of ERK (pERK) correlated with the activity of the GR, and that the expression patterns of pERK and GR were partially overlapping. The downregulation of pERK in the cochlea after RS plus trauma may result from increased activity of GR after restraint, causing GR-induced hearing protection (Tahera et al., 2006c). The possible underlying mechanism of pERK downregulation is a decrease in glutamate-induced exitotoxicity, and inhibition of the generation of the reactive oxygen species in the cochlea after GR activation. Nevertheless, no change in pERK expression was found immediately after RS alone, which suggests that the activity of the ERK pathway is regulated by the complex of general (HPA axis activation) and local (glutamate-induced exitotoxicity and reactive oxygen species) disturbances that occur after acoustic trauma. The reversed effect of RS plus trauma after metyrapone-plus-RU486 treatment on pERK confirms that pERK downregulation occurred in a GR-dependent manner. The decrease in pERK seen after metyrapone-plus-RU486 treatment compared to vehicle may be a result of a decreased Na+ concentration in the endolymph, which has been reported previously (Kim et al., 2009).
P38 MAP kinase is activated in the cochlear nerve by RS in a GR-dependent manner
The activation of p38 MAP kinase by ischemic preconditioning had a protective effect on myocardium (Nagy et al., 2007) and the hippocampus (Nishimura et al., 2003). Prenatal RS induced an upregulation of p38 in the hippocampus of the offspring (Cai et al., 2008), but post-natal restraint did not alter p38 levels in the same brain region (Meller et al., 2003). In the presence of a p38 inhibitor, there was protection against aminoglycoside ototoxicity (Wei et al., 2005) and hypoxia-induced proinflammatory cytokine production in hair cells (Jeong et al., 2005). We found an elevation of p38 in the auditory nerve after RS, suggesting that p38 activation can protect hearing. Table 1 is a summary of the results obtained for all groups compared to vehicle alone. On the other hand, nearly the same levels of p38 MAP-kinase expression were detected in mice pretreated with a GR-antagonist followed by restraint and trauma (Tahera et al., 2006b). This suggests that upregulation of p38 in the auditory nerve can be triggered by GR activation in the absence of acoustic trauma, while acoustic trauma can increase pp38 via GR-independent pathways. Thus, GR can specifically trigger p38 activity in the auditory nerve, but not in cochlear tissues. The activation of p38 MAP kinases by glucocorticoids has been previously shown in vascular smooth muscle cells, myocytes, neutrophils, and primary cultured hippocampal neurons (Kewalramani et al., 2008; Molnar et al., 2008; Qi et al., 2005; Saffar et al., 2008). The glucocorticoid-induced activation of MAP kinases in the auditory nerve may be the result of Ca2+ metabolism and nitric oxide production (Yukawa et al., 2005), or may be a consequence of GR-regulated ion transport in perilymph (Lee and Marcus, 2002). The mechanism of RS-induced protection of the cochlea via the p38 pathway may include the regulation of glutamate-induced transcription (Zepeda et al., 2008), or the activation of antioxidant systems (Yin et al., 2000) by RS prior to acoustic trauma.
Acoustic trauma induces different events in the cochlea that may affect the activity of p38 MAP kinase in a GR-independent manner. For example, it is known that acoustic trauma results in the activation of NMDA receptors that reside on the nerve endings of the spiral ganglion neurons (Pujol et al., 1993). The NMDA-induced activation of p38 in cortical and hippocampal neurons has been previously reported (Moult et al., 2008), and may be a possible mechanism for the p38 activation seen after RS plus trauma in the metyrapone-plus-RU486-treated group. Another cross-link between acoustic trauma and p38 is the induction of reactive oxygen species, which is one mechanism of noise-induced cochlea damage (Ohlemiller et al., 1999; Yamane et al., 1991), and is a potent activator of the p38 MAP kinase pathway (Ito et al., 2006). In the presence of a GR antagonist, two stressful events (RS and trauma) cause the upregulation of pp38, resulting in exacerbated damage to the cochlea, while the single stressor (either RS or trauma) had no effect on pp38 expression in the auditory nerve. We suggest that the damaging effects of RS and trauma were exacerbated when the activity of GR was decreased by metyrapone plus RU486.
JNK-mediated pathway is not activated by RS or acoustic trauma
The JNK-mediated pathway is activated by different types of stressors, including oxidative stress, proinflammatory factors, and acoustic trauma (Matsui et al., 2004; Mendelson et al., 1996; Xia et al., 2000). In-vivo experiments have shown a role of the JNK-mediated pathway in hair cells after acoustic trauma or impulse noise exposure (Murai et al., 2008; Wang et al., 2007), whereas JNK inhibitors prevent trauma-induced JNK activation and hair cell death (Eshraghi et al., 2007; Pirvola et al., 2000; Wang et al., 2003). The treatments used for the above-mentioned studies gave rise to permanent hearing loss, accompanied by hair cell loss and degeneration of spiral ganglion neurons. However, the trauma used in the present study resulted in temporary hearing loss, and did not cause cell death or degeneration of hair cells or neurons. Thus, pJNK upregulation could be attributed to the type of cochlea damage incurred (temporary versus permanent). Moreover, a decrease in pJNK expression in all cell types of the cochlea was demonstrated after less intense acoustic trauma (70–90 dB SPL) (Selivanova et al., 2007). We were not able to confirm this fact, probably because of differences in experimental species, or in the frequency or duration of the acoustic trauma. On the other hand, the time-course is known to be critical for the activity of stress-regulated proteins such as JNK (Murai et al., 2008).
Conclusion
Our data show that the phosphorylated forms of three MAP kinases (JNK, ERK, and p38) are expressed in cochlear tissues, and their levels are not changed after an acoustic exposure that results in a temporary hearing loss. RS or RS followed by acoustic trauma altered the expression of p38 and ERK in the inner ear. Furthermore, the involvement of GR in the regulation of the expression of these MAP kinases after restraint or acoustic trauma was demonstrated for the first time. Taken together, these data partially describe the mechanisms underlying the protective effect of RS on hearing sensitivity. Further elucidation of the interplay between GR and MAP kinases in the cochlea is needed to better understand the potential of glucocorticoid therapy for the prevention and treatment of hearing loss.
Footnotes
Acknowledgments
This work was supported by grants from the Swedish Research Council, AMF Trygghetsförsäkring, Tysta Skolan, and Karolinska Institutet. The authors would like to thank Agneta Viberg for her expert technical assistance.
Author Disclosure Statement
No competing financial interests exist.