
Abstract
Accumulating data suggest that matrix metalloproteinases (MMPs) may be important mediators in the pathophysiology of acute brain injury after trauma or stroke. Here, we test the hypothesis that the endogenous tissue inhibitor of metalloproteinase (TIMP-1) is neuroprotective in vitro and in vivo. For in vitro studies, primary cortical neuronal cultures were subjected to hypoxia and reoxygenation. Treatment with recombinant TIMP-1 protein significantly decreased neuronal death. In vivo studies in models of brain trauma and stroke supported these cell culture results. After controlled cortical impact, 24-h MMP-9 levels were significantly reduced in transgenic mice overexpressing TIMP-1 compared to wild-type mice. And at 7 days post-trauma, brain lesion volumes were also significantly decreased by TIMP-1 overexpression as well. In a model of transient 2-h focal cerebral ischemia, MMP-9 levels were lower in TIMP-1 transgenic mice compared with wild-types. Correspondingly, blood-brain barrier leakage was ameliorated by TIMP-1 overexpression, and 24-h infarction volumes were also reduced. Taken together, these cell culture and in vivo data provide initial proof-of-principle that TIMP-1 is neuroprotective against traumatic and ischemic brain injury in mice.
Introduction
M
It has been previously demonstrated that knockout mice lacking MMP-9 were protected against traumatic brain injury (Wang et al., 2000) as well as focal and global cerebral ischemia (Asahi et al., 2000, 2001; Gidday et al., 2005; Lee et al., 2004). If MMPs promote brain injury, then inhibition of these proteases should be beneficial. However, development of specific MMP inhibitors can be challenging, and untitrated blockade of pathophysiologic pathways with exogenous compounds may sometimes lead to unanticipated side effects. In this regard, we decided to ask whether an endogenous approach may also be useful. MMPs do not work in isolation. Instead, these proteases function as part of a complex network in dynamic balance with a class of endogenous inhibitors called tissue inhibitors of metalloproteinase (TIMPs) (Candelario-Jalil et al., 2009). In the present study, we use a combination of cell culture and in vivo experiments to ask whether the potent MMP-9 inhibitor, TIMP-1 is neuroprotective. Our data demonstrate that (i) in primary neuronal cultures, recombinant TIMP-1 reduced neuronal death after hypoxic injury, (ii) in a controlled cortical impact model, TIMP-1 overexpressing transgenic mice were protected against cortical damage, and (iii) in a focal cerebral ischemia model, TIMP-1 transgenic mice were protected against infarction and blood-brain barrier damage. These data provide proof-of-principle that TIMP-1 may be broadly neuroprotective against acute brain injury.
Methods
Traumatic and ischemic brain injury models
All experiments were performed following an institutionally approved protocol in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments were performed with halothane (1–1.2%) under spontaneous respiration in an air/oxygen mixture. Rectal temperature was maintained at 37.5°C with a thermostat-controlled heating pad. For trauma, a standard controlled cortical impact model was used following standard methods (Mori et al., 2002; Wang et al., 2000). In a stereotactic frame, a 5-mm craniotomy was performed over the right parietal cortex between bregma and lambda, a 3-mm flat-tipped impactor was placed on the dural surface at a 20° angle, and injury was induced using 4 m/s impact velocity, 1-mm impact depth, and 150-ms impact dwell time. Transgenic TIMP-1 mice were compared with matching wild-type mice. All mice survived the trauma procedure. At 7 days after injury, mice were deeply anesthetized, transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde, and brains were rapidly removed and sectioned with a freezing microtome into 20-μm-thick coronal slices. Every 25th section was mounted onto a glass slide and stained with 0.1% cresyl violet. Histological lesion areas were quantified with a standard computer-assisted image analysis program, and lesion areas were integrated to obtain total lesion volumes. Lesion delineation of the necrotic cavitation post-trauma was performed blindly using criteria established in previous studies in this mouse model (Mori et al., 2002; Wang et al., 2000). For focal cerebral ischemia, the standard intraluminal filament model was used (Tsuji et al., 2005). Silicon-coated 7.0 monofilaments were advanced via the internal carotid arteries to occlude the middle cerebral artery. To induce reperfusion, filaments were withdrawn after 2 h. Stable ischemia and reperfusion were confirmed with laser Doppler flowmetry. Because we used a transient ischemia model, mortality and exclusions were low. Consistent with our typical experience with this model, one mouse was excluded because of filament puncture and hemorrhage, four were excluded because laser Doppler levels did not drop below the pre-specified 30% for adequate ischemia, and two died within the first 24 h. There were no obvious differences between mouse strains. To assess ischemic injury, brains were removed at 24 h and stained with triphyenyl tetrazolium chloride. To assess blood-brain barrier leakage, the standard Evans blue leakage assay was used (Aoki et al., 2002; Asahi et al., 2001). Transgenic mice, overexpressing human TIMP-1 under the metallothionein-1 promoter, were obtained from Rama Khokha, Ontario Cancer Center, Toronto, Canada. Endogenous levels of TIMP-1 have been previously established to be elevated at least threefold over transgene-negative littermates (Kruger et al., 1998).
Gelatinase zymography
Brains were removed after transcardial perfusion with ice-cold PBS (pH 7.4), frozen immediately on dry ice, and stored at −80°C. For further analysis, brain samples were homogenized in lysis buffer that included protease inhibitors on ice. After centrifugation, supernatant was collected, and total protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA). Prepared protein samples were loaded and separated by 10% Tris-glycine gel with 0.1% gelatin as substrate. After separation by electrophoresis, the gel was renaturated and then incubated with developing buffer at 37°C for 24 h. After developing, the gel was stained with 0.5% Coomassie Blue R-250 for 30 min and then destained. Data were quantified with image densitometry.
Primary neuronal culture model
Primary cortical neurons were established followng standard protocols. Briefly, mouse neurons were cultured from E17 embryonic cortices, and isolated cells were plated onto poly-
Statistical analysis
Data were expressed as mean ± standard deviation (SD), and statistical analysis was performed by standard analysis of variance (ANOVA), followed by Tukey's test between individual groups. Differences for which p was <0.05 were considered significant.
Results
Primary cortical neurons were subjected to 6 h of hypoxia, followed by 18 h of reoxygenation. As expected, this hypoxia-reoxygenation insult was neurotoxic. Wild-type neurons showed about 45% cell death, as measured by a standard LDH release assay. Neurotoxicity was significantly reduced to approximately 22% when cultures were simultaneously treated with 500 ng/mL of recombinant TIMP-1 (Fig. 1).
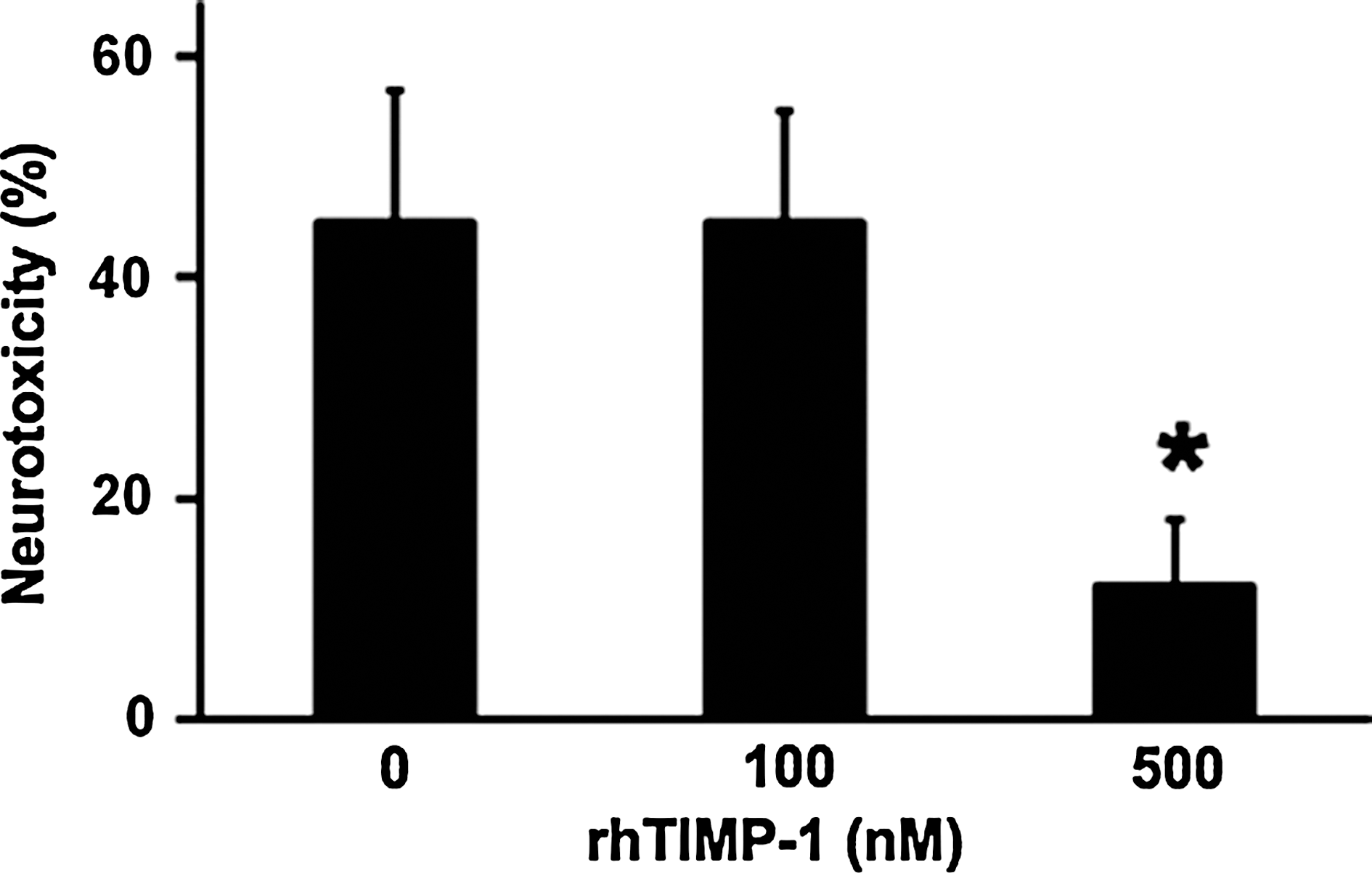
Lactate dehydrogenase (LDH) release in primary mouse cortical neurons after 8 h of hypoxia, followed by 16 h of reoxygenation. Neurotoxicity was significantly ameliorated by treatment with recombinant human tissue inhibitor of metalloproteinase—1 (TIMP-1) at 500 nM concentrations. *p < 0.05; n = 4 independent experiments performed in duplicate.
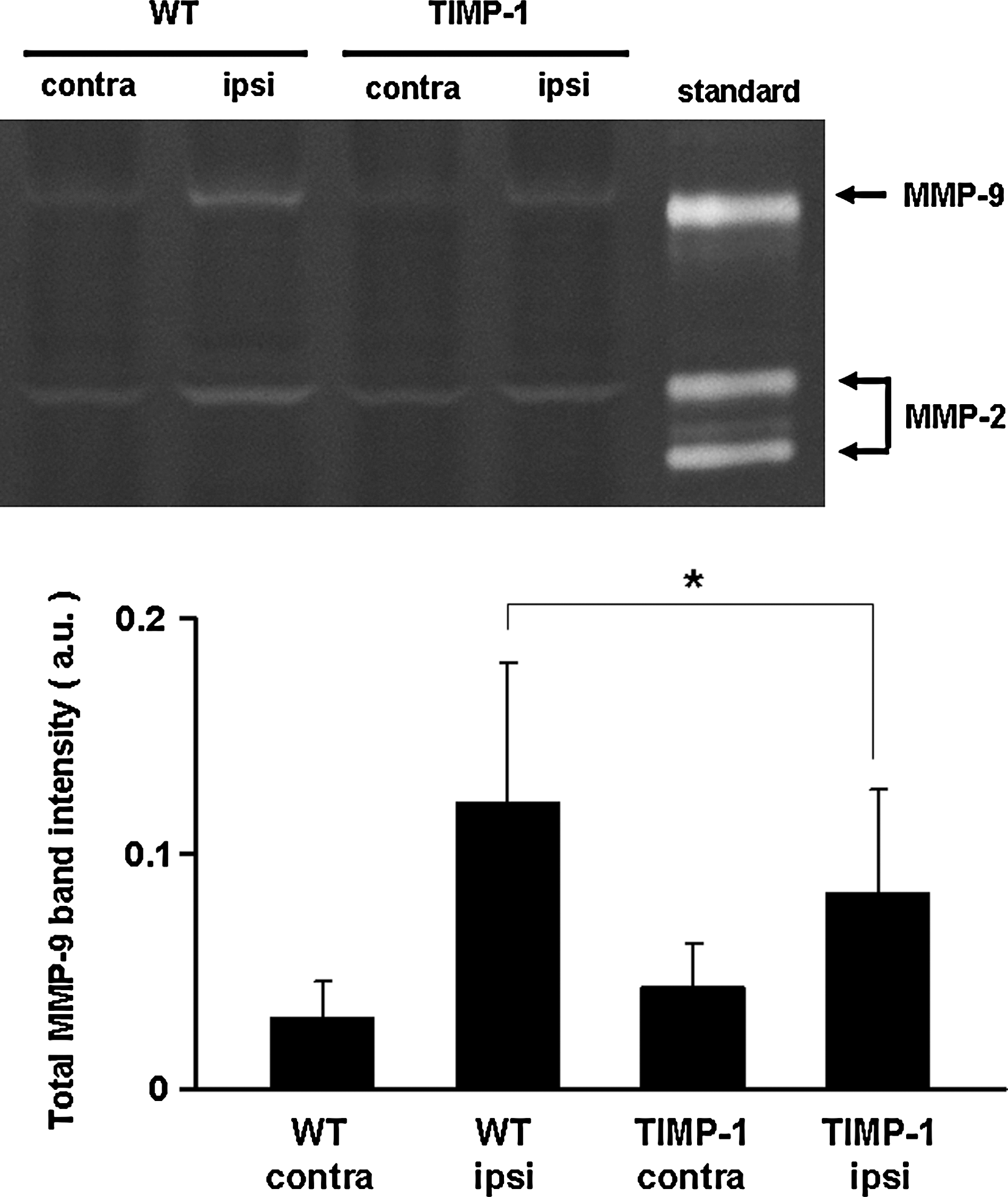
We next turned to in vivo models of brain injury. First, a standard model of traumatic brain injury was implemented using controlled cortical impact in wild-type mice and TIMP-1 overexpressing transgenic mice. At 24 h after cortical trauma, gel zymography showed that brain MMP-9 levels were lower in transgenic mice compared to wild-type mice, confirming the MMP-suppressing actions of TIMP-1 (Fig. 2). As expected, at 7 days after injury, brain lesions were apparent as cortical areas of parenchymal injury and cavitation. Lesion volumes in transgenic TIMP-1 overexpressing mice (5.8 ± 0.5 mm3) were significantly smaller compared to those in wild-type mice (6.9 ± 0.9 mm3; Fig. 3).

Gel zymograms of tissue homogenates at 24 h after brain trauma. (

Traumatic lesion volumes at 7 days after controlled cortical impact. Lesion volumes were significantly smaller in tissue inhibitor of metalloproteinase—1 (TIMP-1) transgenic brains (n = 10), compared with wild-type brains (n = 8). *p < 0.05.
Experiments were next performed in a model of transient focal cerebral ischemia. Upon arterial occlusion, cerebral perfusion dropped below 25% in all mice; there were no differences in TIMP-1 transgenics versus wild-types (Table 1). Systemic parameters remained within normal range with no differences between wild-type or knockout mice (Table 2). To assess effects of TIMP-1 overexpression on MMPs, gel zymography was performed on brain sections and homogenates at 24 h after ischemic onset. These data confirmed that MMP-9 levels were significantly decreased by TIMP-1 overexpression in ischemic brain homogenates (Fig. 4). At 24 h after a 2-h transient occlusion of the middle cerebral artery, ischemic lesions were apparent in cortex and basal ganglia, as expected. Lesion volumes were significantly reduced in transgenic mice (45.9 ± 17.1 mm3) compared with wild-type mice (116.2 ± 16.1 mm3; Fig. 5A).
Gel zymograms of tissue homogenates at 24 h after 2 h of transient focal cerebral ischemia. (
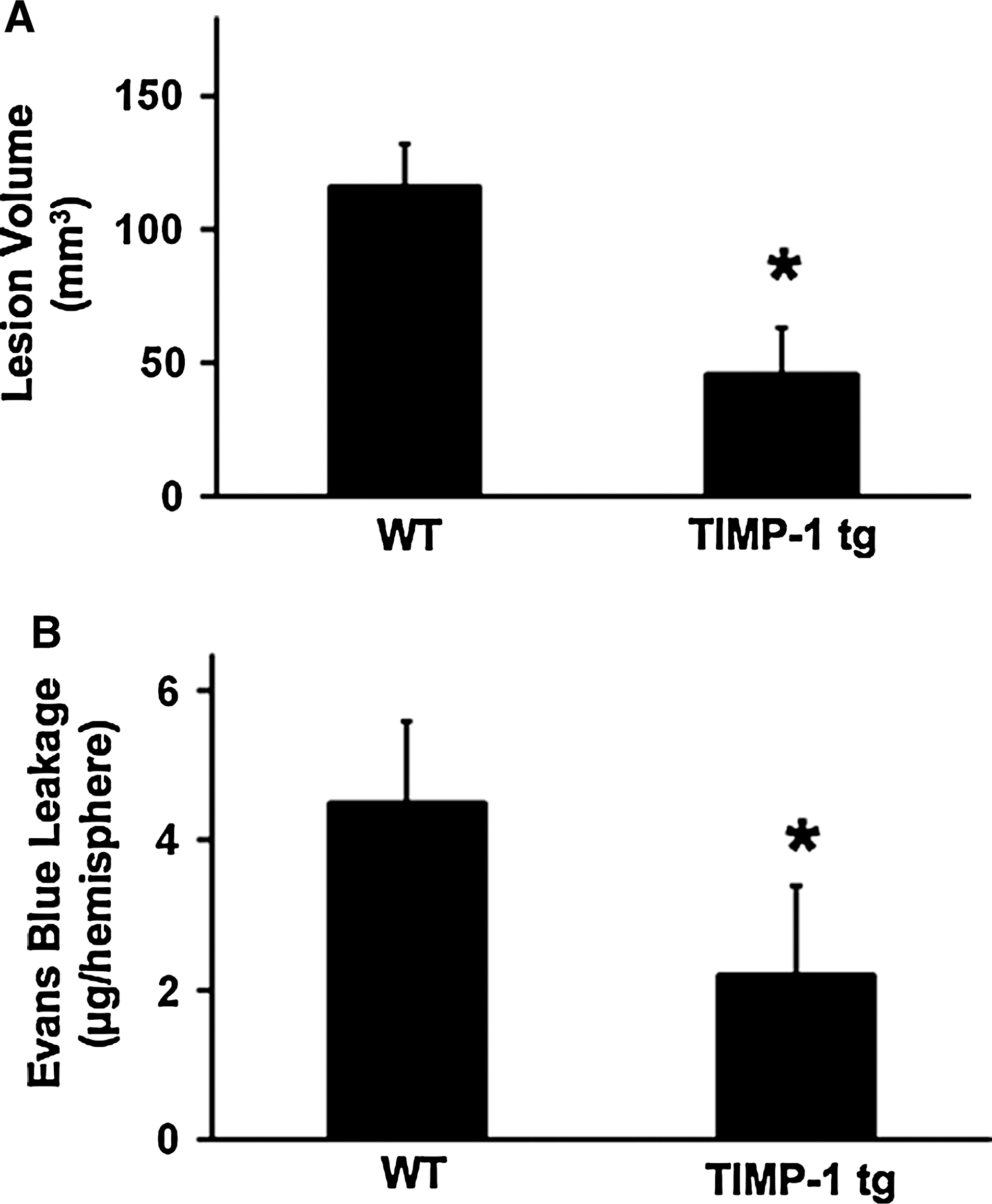
Brain injury after focal cerebral ischemia. (
WT, wild-type; TIMP-1, tissue inhibitor of metalloproteinase–1; tg, transgenic.
WT, wild-type; TIMP-1, tissue inhibitor of metalloproteinase–1; tg, transgenic; BP, blood pressure.
Finally, we assessed the effects of overexpressing TIMP-1 on ischemic blood-brain barrier injury. Evans blue dye leakage at 24 h after 2-h transient focal cerebral ischemia was significantly reduced in TIMP-1 transgenics (2.2 ±1.2 μg/hemisphere) compared with wild-type mice (4.5 ±1.1 μg/hemisphere; Fig. 5B).
Discussion
Increasingly, clinical evidence suggests that MMPs are elevated in a wide range of central nervous system (CNS) disorders. In particular, the inducible gelatinase MMP-9 has been extensively investigated. MMP-9 protein is detected in brain tissue from ischemic and hemorrhagic stroke patients (Rosell et al., 2006; Tejima et al., 2007). MMP-9 appears to be elevated in brain tissue (Asahina et al., 2001; Thirumangalakudi et al., 2006) and cerebrospinal fluid (CSF) (Lorenzl et al. 2003) of Alzheimer's disease patients. Similar CSF elevations in MMP-9 have also been described for patients with vascular dementia (Adair et al., 2004; Rosenberg et al., 2001b). Some of these MMP-9 signals may also be detectable in blood and plasma of patients with cerebrovascular disease and neurodegeneration, thus raising the possibility that they may additionally serve as surrogate biomarkers of disease (Montaner, 2006). The limitation remains, however, that MMP-9 may also be a nonspecific inflammatory marker. These clinical findings cannot truly tell us whether elevated MMP-9 contributes to disease or merely reflects ongoing neuronal injury and inflammation.
In experimental model systems, data to support a causal role for MMPs (and MMP-9 in particular) may be easier to interpret. Chemical inhibitors of MMPs appear to reduce tissue damage after brain trauma (Shigemori et al., 2006; Wang and Tsirka, 2005) and cerebral ischemia (Asahi et al., 2000; Gu et al., 2005; Jiang et al., 2001; Lee et al., 2004; Romanic et al., 1998). Furthermore, knockout mice lacking the MMP-9 gene are protected against traumatic brain injury, focal cerebral ischemia, and global cerebral ischemia (Asahi et al., 2000, 2001; Gidday et al., 2005; Lee et al., 2004; Wang et al., 2000). In this study, we provide further evidence to support a mechanistic role for MMPs. If MMP gene knockout protects brain, then it is reasonable to hypothesize that overexpressing endogenous MMP inhibitors should decrease injury. Our data showed that (i) treatment with recombinant TIMP-1 decreased neurotoxicity in primary cortical neurons after hypoxic injury, (ii) TIMP-1 overexpressing transgenic mice were protected against traumatic brain injury, and (iii) infarct volumes and blood-brain barrier leakage were reduced in TIMP-1 transgenic mice after focal cerebral ischemia. Taken together, our findings support the central idea that MMPs play critical roles in neuronal death after both brain trauma and stroke. These findings are consistent with the overall idea that, in conditions of brain disease, tissue damage, and inflammation, there may be an imbalance in the dynamic equilibrium between MMPs and TIMPs, so that any way of enhancing the inhibitor side of the equation should be beneficial (Sellner and Leib, 2006; Waubant et al., 2003). We previously showed that infusion of recombinant TIMP-1 was effective in a rat model of trauma-induced neuropathic pain (Kawasaki et al., 2008). Other groups showed that viral delivery of TIMP-1 or TIMP-2 were neuroprotective in rat models of focal cerebral ischemia (Baker et al., 2007; Magnoni et al., 2007). Furthermore, TIMP-1–deficient knockout mice were found to be more vulnerable to cerebral ischemia (Fujimoto et al., 2008).
Taken together, our findings here suggest that raising TIMP-1 levels may be neuroprotective in vitro and in vivo. However, there are several caveats. First, TIMP-1 can inhibit a wide range of MMPs, even though it has been described as being particularly potent against MMP-9 (Gardner and Ghorpade, 2003). Our internal controls (i.e., decreased ischemic damage in MMP-9 knockout mice [data not shown]) suggested that MMP-9 played a critical role in our model systems. But it is acknowledged that MMPs operate in a broad network-like manner. Further studies are needed to determine how overexpression of TIMP-1 disrupts not just gelatinases such as MMP-9 and MMP-2 but other proteases in the MMP family. A second caveat relates to the question of neuronal death mechanism. MMPs can amplify cerebral injury via neuroinflammation (Rosenberg, 2002), and disruption of cell-matrix homeostasis may mediate anoikis-like cell death (Gu et al., 2002, 2005; Lee and Lo, 2004). What type of specific cell death pathways are interrupted by TIMP-1 remains to be fully elucidated. A third limitation involves the difficulty in unequivocally proving mechanisms and causality. MMP-9 was reduced in our TIMP-1 overexpressing models. But it is possible that decreased MMP-9 may simply reflect a lesser degree of injury due to completely non-MMP-related reasons. MMPs are upregulated by oxidative stress and inflammatory cytokines. So any nonspecific amelioration of these pathways can result in lower levels of MMP-9 observed here. Broadly speaking, the entire TIMP family is now recognized to possess many other roles beyond metalloprotease inhibition per se (Chirco et al., 2006; Stetler-Stevenson, 2008). For example, TIMP-1 can directly suppress apoptosis (Liu et al., 2003), in part by binding onto CD63 receptors and interacting with integrin signaling pathways (Jung et al., 2006). Whether these signals influence outcomes in our models should be examined further. Finally, our primary data comprise acute morphological endpoints, and we did not assess long-term behavioral outcomes. Whereas TIMP-1 may be beneficial in the acute phase by decreasing neuronal injury, it is unclear what happens as brain tissue recovers. Emerging data now suggest biphasic roles for MMPs in brain injury (Zhao et al., 2007). Inhibition of MMPs during acute stages of cerebral ischemia or trauma may be beneficial, but targeting MMPs during the delayed stages may be detrimental, as it might interfere with neurogenesis, neurovascular remodeling, and matrix-trophic signaling in recovering brain tissue (Falo et al., 2006; Lee et al., 2006; Reeves et al., 2003; Wang et al., 2006; Zhao et al., 2006). Some have suggested that TIMP-1 can also interfere with angiogenesis (Guedez et al., 2001), thus raising the problematic possibility that it might worsen neurovascular recovery after brain injury. More detailed studies are required to examine how increasing TIMPs may influence the delicate balance between acute injury versus delayed plasticity and recovery (Lo, 2008).
In conclusion, our data here demonstrate that TIMP-1 can ameliorate hypoxic neuronal cell death in vitro and decrease traumatic and ischemic brain damage in vivo. MMPs play a critical role in stroke, trauma, and neurodegeneration. It is difficult to develop specific chemical inhibitors of MMPs. This proof-of-principle study suggests that using recombinant TIMP proteins or other strategies to upregulate endogenous inhibitors of MMPs may provide adjunct therapeutic approaches for acute brain injury. However, translating experimental ideas into positive outcomes in stroke and trauma patients is extremely challenging, and this TIMP-1 hypothesis remains to be validated in a true clinical situation.
Footnotes
Acknowledgments
This research was supported in part by grants from NINDS and a Bugher award from the American Stroke Association.
Author Disclosure Statement
No competing financial interests exist.