
Abstract
Shock-wave exposure from improvised explosive devices (IEDs) has been implicated as a possible contributing factor to neurological impairment reported in combat veterans. However, evidence-based substantiation of this implication, particularly for low-level exposure in the absence of external signs of trauma, remain elusive. Accordingly, we constructed an open-ended shock tube producing a short-duration, low-amplitude shockwave. Low-level (11.5 kPa static overpressure) complex shock-wave exposure in rats resulted in no histological evidence of lung injury. By contrast, delayed cytoskeletal proteolysis of αII-spectrin was detected in the cortex and hippocampus by 12 h post-injury. Cell death was minimal and localized predominantly in the corpus callosum and periventricular regions. These regions, with presumably different density interfaces, exhibit biological responses to shockwaves consistent with interface turbulence described by Richtmyer-Meshkov instability. Evoked compound action potential (CAP) recordings from the corpus callosum showed a significant increase in the duration of CAP responses at 14 and 30 days post-injury, and a gradual depression in the unmyelinated fiber amplitude. Shielding the head attenuated αII-spectrin cytoskeletal breakdown, thus directly implicating low-level shock-wave exposure as a cause of brain injury in the rat. Despite anatomical and scaling differences in rats compared to humans, the results suggest the potential for undiagnosed traumatic brain pathologies occurring in combat veterans following shock-wave exposure.
Introduction
Blast injuries are categorized into four components (DePalma et al., 2005). First, primary blast trauma refers to injuries sustained by exposure to the shockwave generated by an explosive detonation. In an open-field blast the leading shock front is characterized by an instantaneous increase in ambient pressure, heat, and density, followed by a prolonged negative pressure phase arising from the trailing expansion fan. From a pathobiological perspective, exposure to high primary blast overpressure typically causes injury to gas-filled organs (e.g., lungs and bowels), and possible rupture of the tympanum (Mayorga, 1997). It is only recently that the potential impact on neurological function has been addressed in detail. It is important to note that these injuries can occur in the absence of visible external injuries, making diagnosis difficult. Next, secondary blast trauma refers to injuries sustained due to penetration of high-velocity debris following detonation. Third, tertiary blast injuries are caused by the blast wind trailing the primary shock front. Blast winds can be sufficiently powerful to result in limb amputation and total body displacement. Finally, quaternary blast injuries are sustained due to high heat and noxious chemical exposure associated with the conflagration following explosive detonations.
In the context of modern combat casualty care, a prime concern is the potential but unsuspected injury sustained by the brain in situations in which sufficient distance or body protection results in occult injury. Blast models used to study the pathophysiological response to shockwaves have included the use of air-driven shock tubes (Bowen et al., 1968; Cernak et al., 1996; Chavko et al., 2007; Gorbunov et al., 1997; Long et al., 2009), explosives-driven shock tubes (Bauman et al., 2009), small- and large-scale charges (Bowen et al., 1968; Garner et al., 2009; Kaur et al., 1997a, 1997b; Nakagawa et al., 2008; Saljo et al., 2000), and firing of weapons with explosive discharge (Saljo et al., 2009a). The majority of small animal pre-clinical studies to date have emphasized high levels of overpressure, with the exception of recent work by Saljo and colleagues (Saljo et al., 2009b). The recent appreciation of the pathophysiological mechanisms in the brain attributable to blast is demonstrated by the numerous methodological approaches taken to address the issue. Reported overpressures used in a variety of mammalian species have ranged from 10 kPa (Saljo et al., 2009b) to as high as 538 kPa (Bauman et al., 2009), with a range of pressures and durations reported in between.
In this regard we have developed a small-scale open-ended shock tube designed specifically for testing low-level overpressures in small rodents. The shock tube produces shockwaves of shorter duration (microsecond duration rather than millisecond), and lower amplitude than previously described models, but it is also capable of producing sufficient overpressure to cause significant barotrauma. We examined the distribution of terminal-deoxy-transferase-mediated dUTP nick end labeling (TUNEL) at 24 and 72 h post-injury in the cortex, hippocampus, and corpus callosum, to ascertain regional cellular vulnerability. Given the higher density of TUNEL expression in the periventricular white matter and corpus callosum relative to the cortex or hippocampus, we subsequently examined expression of the axonally-expressed proteins neurofilament protein 200 (NF200) and alphaII-spectrin (αII-spectrin). Immunoblot data, including expression of myelin protein 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), were complemented with electrophysiologic compound action potential (CAP) recordings from the corpus callosum in acute brain slices of shock-wave exposed rats. Collectively the results support the notion that low-level shock-wave exposure can alter normal white matter conduction velocities, and induce persistent alterations of the expression levels of white matter cytoskeletal proteins under conditions of minimal cell death.
Methods
All surgical procedures were performed in accordance with guidelines established by the Animal Care Committee at St. Michael's Hospital in accordance with the standards set by the Canadian Council on Animal Care. A total of 124 adult male Sprague-Dawley rats were used for this study.
Primary blast shock-wave model
The open-ended shock tube device consisted of a cylindrical driver chamber (6×1.5-cm diameter) pressurized through a pin-hole opening fed from a pressurized reservoir containing nitrogen gas (Fig. 1A). The base of the driver chamber was sealed through a threaded mount with a diaphragm made of aluminum. Various diaphragm thicknesses were evaluated, resulting in a range of overpressures being generated as a function of the driver pressure required to rupture the diaphragm. Data for this study were collected using a diaphragm with a thickness of 0.27 mm, with the exception of some experiments in which a 0.48-mm diaphragm was used to generate higher overpressures at 35 and 66 kPa. The entire driver system was mounted on a vertically ascending/descending frame, that allowed exposure to shockwaves over a range of pressures by varying the distance to the source. The 0.27-mm-thick aluminum bursting diaphragm (1.5 cm diaphragm opening) ruptured at approximately 4.48 MPa.
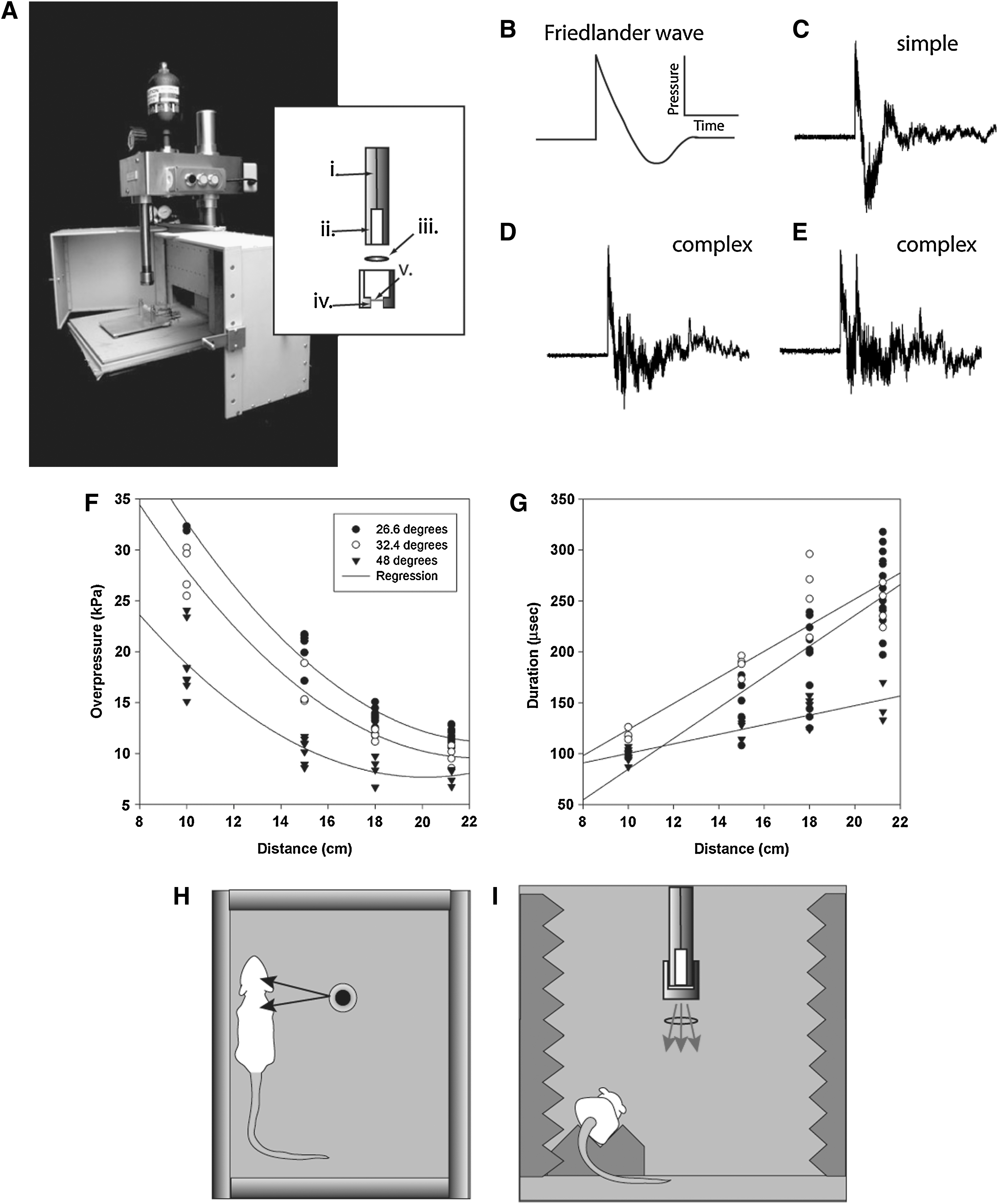
(
In order to validate and characterize the shockwave being produced, static pressure measurements were recorded at 26.6°, 32.4°, and 48.2° degrees from the vertical, at distances of 10, 15, 18, and 21.5 cm from the nozzle opening for each off-axis angle. Static pressure measurements were made with a custom-built swivel-mount pencil probe fitted with a DPX101 high-frequency piezoelectric pressure sensor mounted perpendicular to the length of the probe (Omega Engineering, Laval, Quebec, Canada). The pressure sensor was connected to an ACC-PSI power source with output to an oscilloscope for data acquisition. Static pressure waveforms were consistent in static locations between measurements. However, different locations resulted in different waveforms. For example, complex waves with at least two overpressure peak events were determined to be caused by floor reflections, and occurred at more obtuse angles, and with closer proximity to the chamber floor. The second overpressure peak could be reduced with the introduction of sound-reduction foam below the sensor (data not shown), suggesting a primary contribution of the reflected wave. Simple waves displayed the characteristics of a Friedlander waveform, having both an instantaneous positive overpressure and an extended underpressure component (Fig. 1B and C). Complex waves ranged from decaying overpressure peaks (Fig. 1D), to dual peaks with similar amplitudes (Fig. 1E). The resultant data were plotted as functions of overpressure versus distance, and overpressure duration versus distance (Fig. 1F and G). The fitted exponential decay curve for overpressure as a function of distance (f=y0+a*exp(−b * x); R2=0.95 for 26.6°, R2=96.4 for 32.4°, and R2=0.94 for 48.2°) was consistent with the pattern of decrease in shock strength described for three-dimensional expansion in an open-ended shock tube (Sloan and Nettleton, 1975). Importantly, the duration of the overpressure event in our model was scaled down to the order of microseconds, rather than milliseconds, as has been recorded in actual explosive detonations (Bauman et al., 2009; Champion et al., 2009). Duration of the incident shockwave increased linearly with distance, which was in accordance with Goodman's report on free-air blast data (Baker, 1973). Pressure measurements at acute angles (e.g., 26.6° and 32.4°) to the vertical axis had higher overpressure amplitudes than equidistant measurements 48.2° from the vertical. This would suggest a shock front with higher amplitude closer to the vertical axis, and diminishing shock strength at the periphery. Collectively, these results are consistent with shock-wave characteristics as described in previous open-ended shock-tube experiments (Yu and Gronig, 1996).
Computational flow dynamic (CFD) modeling indicated that the jet stream and mach disk arising from the vented driver gas were localized to a cylindrical region with an approximate 6-cm radius (data not shown). Static pressure diagrams from the CFD model suggested that the diffracted wave arising off-axis to the nozzle would provide the closest approximation to explosive primary blast-wave overpressure conditions, while minimizing the confounding effects of the mach disk and jet stream. Accordingly, ketamine-anesthetized (100 mg/kg; Bimeda-MTC, Cambridge, Ontario, Canada) adult male Sprague-Dawley rats (350–400 g) were placed 8 cm off-axis and on a sound-reducing foam bed to minimize exposure to wave reflections from the floor (Fig. 1H and I). The rat's head and body were equidistant from the nozzle opening. The angle of exposure for low-level exposure was at 22.3° and 21 cm from the nozzle opening, resulting in a measured overpressure of 11.5 kPa, with a consistent complex decaying waveform represented in Figure 1D.
Bronchiolar lavage and lung histology
In order to validate that the shockwave was sufficiently titrated to result in no lung injury, we assessed lung tissue histologically for evidence of altered morphology (n=4 at 11.5 kPa; n=4 at 35 kPa; n=4 at 66 kPa; n=4 for naïve) at 24 h post-injury. Bronchiolar lavage (BAL) was also performed at 24 h, and the bronchiolar lavage fluid (BALF) was examined for evidence of increased inflammatory cell populations. Briefly, under ketamine and xylazine anesthesia (10 mg/kg; Bayer Inc., Toronto, Ontario, Canada), the rats were exsanguinated via the descending aorta. The trachea was cannulated and the lungs were flushed three times with 0.9% saline at a volume of 33 mL/kg. The BALF was spun at 2000 g for 10 min. The pellet fraction was resuspended in 6 mL of 0.1 M phosphate-buffered saline (PBS). A 100-μL sample was transferred to a slide using a cytospin, and stained with HEMA-3 (Sigma-Aldrich, Oakville, Ontario, Canada), according to the manufacturer's protocol for identification of inflammatory cell type based on size and nuclear morphology (Taniuchi et al., 2009). We quantified the number of macrophages in normal and 11.5-kPa shock-wave-exposed BALF. The cells were quantified with a 10×objective on a Nikon 90i light microscope.
Following BAL, the lungs were filled with 4% paraformaldehyde (33 mL/kg) for 5 min, then extracted and post-fixed in paraformaldehyde overnight. The lungs were then paraffin embedded and the right middle lobe was sectioned at 10 μm for histological processing. Lung sections were stained with hematoxylin and eosin (H&E) for morphological assessment. Images were captured with a 20×objective on the light microscope. Given the length of time required for BAL preparation and the potential for protein degradation, the brains from the animals used in the lung barotrauma experiments were not used for immunohistochemistry or Western blot analysis.
Immunohistochemistry
Rats were anesthetized with a mixture of ketamine and xylazine, then were transcardially perfused at 24 and 72 h post-injury with 0.9% saline, followed by 250 mL of 4% paraformaldehyde (n=5 per time point). The extracted brains were post-fixed overnight in 4% paraformaldehyde with 0.5 M acetate solution prior to paraffin embedding. Paraffinized coronal brain sections 10 μm thick were used for fluorescent immunohistochemistry. For fresh frozen processing, the brains were cryoprotected in 30% sucrose in PBS overnight at 4°C. Cryoprotected brains were snap frozen in Cryomatrix (Fisher Scientific, Ottawa, Ontario, Canada), and sectioned on a cryostat at a thickness of 12 μm. Frozen sections were used for immunoperoxidase staining.
Naïve and shock-wave-exposed brains were probed for calpain-mediated breakdown of spectrin using a custom produced antibody, PAC2375 (Pacific Immunology, Ramona, CA), generated from a slightly modified previously published and validated peptide sequence CQQQEVY (Roberts-Lewis et al., 1994). Our peptide sequence included a carbon spacer and a cysteine moiety at the tyrosine residue, with a final sequence of QQQEVY-Ahx-C. Peroxidase activity in frozen sections was quenched with 3% H2O2 in methanol for 20 min. Sections were permeabilized for 25 min with 0.1% Triton-X. Blocking of sections was performed in 0.1 M PBS containing 10% normal goat serum. Primary antibody incubation of PAC2375 was performed overnight at a dilution of 1:20,000. Biotinylated goat anti-rabbit secondary antibody was used for positive signal detection in conjunction with an ABC kit and Vector VIP immunoperoxidase substrate (Vector Laboratories, Burlington, Ontario, Canada).
Double-label fluorescence immunohistochemistry for confirmation of PAC2375-expressing cells was performed with monoclonal anti-NeuN (Sigma-Aldrich) for identification of neurons. Antigen retrieval was performed by incubating sections in sodium citrate buffer and microwave heating. Primary antibody incubations were performed in 10% normal goat-serum in 0.1 M PBS overnight at 4°C at a dilution of 1:1000 for NeuN, and 1:20,000 for PAC2375. Secondary antibody incubation was performed sequentially with Alexa Fluor 488 goat anti-mouse for detection of NeuN, and Alexa Fluor 555 goat anti-rabbit antibodies for detection of PAC2375 (Invitrogen, Burlington, Ontario, Canada), for 1 h at room temperature in blocking solution at a dilution of 1:1000. Three washes were made in 0.1 M PBS between each step. Hoechst was used as a nuclear counter-stain. Negative controls were run as described, with the exception of omitted primary specific antisera.
Quantitative immunohistochemistry
Sections were sampled from 4 animals per treatment group at 24 and 72 h post-injury. The sections were sampled at approximately −2.5 mm and −4.5 mm to the bregma, based on coordinates from Paxinos and Watson (Paxinos and Watson, 1998). Two sections per sampling coordinate for a total of 4 sections per animal were obtained for quantification of TUNEL. An in situ fluorescein cell death kit (Roche Applied Science, Laval, Quebec, Canada) was used for TUNEL assays according to the manufacturer's protocol. In each sampled section, the total number of TUNEL-positive cells was quantified from the entire corpus callosum, cortex, hippocampus, and periventricular lining of the lateral ventricles (up to a depth of 30 μm radiating from the ventricles). The number of TUNEL-positive cells for each region was averaged over the four sampled sections as a representative value for each region per animal. That value was then expressed as a ratio of TUNEL-positive cells to the total surface area of the region quantified.
Cellular density in the sampled regions was assessed through quantification of Hoescht-positive nuclei in a 20×sampling field. Nine sampling fields per brain region were imaged and quantified.
Western blotting
Brain tissue samples were collected from both hemispheres of the cerebral cortex and hippocampus from −2.5 mm to −4.5 mm from the bregma. The majority of the corpus callosum was extracted along with the hippocampal tissue, although we cannot definitively rule out some contamination in the cortex samples. Post-injury time points assessed were 6, 12, 24, and 72 h, and 7 and 14 days post-injury (n=6 per time point). Samples were homogenized in lysis buffer containing protease inhibitors (50 mM Tris-HCl, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 μg/mL pepstatin). Protein quantification was determined by the modified Lowry method (Peterson, 1977). Samples were normalized for equal loading (40 μg/lane), and were electrophoresed on 7.5% or 12% SDS-PAGE gels, and transferred overnight to nitrocellulose membranes. Blocking of membranes was performed in 5% non-fat milk blocking solution for 1 h at room temperature. Immunoblots were probed for calpain-mediated breakdown of spectrin, PAC2375. Primary incubations were performed overnight at 4°C at a dilution of 1:15,000 in blocking solution. Secondary antibody incubation was performed for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse (1:3000 dilution) secondary antibodies. Rabbit polyclonal anti-ERK1,2 antibody (Sigma-Aldrich) was used as a loading control (1:15,000 dilution), and visualized with HRP-conjugated goat-anti-rabbit secondary antibodies (1:3000 dilution). Immunoblotting of intact spectrin and the 145/150 and 120 breakdown products was performed with a mouse monoclonal antibody (Millipore, Billerica, MA), incubated at a dilution of 1:20,000 overnight at 4°C. The expression of the heavy-chain neurofilament protein NF200 was examined using a monoclonal anti-NF200 antibody (Sigma-Aldrich) that recognizes phosphorylated and non-phosphorylated forms of the protein. Primary antibody incubation of NF200 was performed at a dilution of 1:1000 at 4°C in blocking solution. Expression of the myelin-associated protein CNPase was examined at 24 and 72 h post-injury with anti-CNPase (Millipore) at a concentration of 1:1000 in blocking solution. Washes in three changes of TBST were performed between incubations. Bands were visualized using a chemiluminescence kit (Perkin Elmer, Waltham, MA), and exposure to x-ray film. Blots were scanned at 300 dpi and densitometry analysis was performed with ImageJ software (
Corpus callosum electrophysiology
Evoked CAP were recorded from the corpus callosum as previously described (Baker et al., 2002). Recordings were made from naïve rats (n=8) at 1, 3, 7, 14, and 30 days post-injury (n=8–9 per time point). In brief, anesthetized rats were decapitated, the brains were extracted in iced artificial cerebrospinal fluid (aCSF) containing 124 mM NaCl, 5 mM KCl, 1.25 mM Na2HPO4, 2 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3, and 20 mM D-glucose, bubbled with a gaseous mixture of 95% O2 and 5% CO2. Four to five recordable coronal brain slices (500 μm thick) were obtained from each rat using a vibratome, and stabilized at room temperature for a minimum of 90 min prior to recording. Electrical stimulation of the corpus callosum was performed using a bipolar tungsten electrode placed over the corpus callosum 750 μm from the recording electrode. Stimulation pulses of constant current (0.1–1.5 mA, 0.1 msec) were generated by a Grass S88 stimulator (Grass Instruments, West Warwick, RI), and delivered through an isolation unit every 30 sec. The evoked CAP was recorded through a glass microelectrode (resistance 2–3 MΩ) filled with 150 mM NaCl, using an Axopatch 200B amplifier, and data were stored and analyzed with pCLAMP 10 software (Molecular Devices, Sunnyvale, CA). The unit of measurement for CAP analysis was the number of animals by averaging CAP values from the slices obtained from a single animal. The maximum evoked CAP amplitude for the myelinated and unmyelinated fibers was calculated using the average of the initial hyperpolarized peak to depolarization, and peak to hyperpolarized peak. Duration of the CAP response was calculated using the maximum CAP response values, by examining the hyperpolarized peak-to-peak response of the CAP waveform.
Statistical analysis
BALF macrophage counts were analyzed using a Student's t-test. TUNEL quantification was analyzed using a one-way analysis of variance (ANOVA), with a Student Newman-Keuls (SNK) post-hoc test. Heavy neurofilament Western blot densitometry analysis was performed using a one-way ANOVA and Dunn's post-hoc test. Electrophysiological data were analyzed using a one-way ANOVA with a SNK post-hoc test. Statistical significance was set at p≤0.05. Errors are shown in the graphs as standard error of the mean (SEM).
Results
Establishment of sub-pulmonary barotrauma shockwave levels
To address the issue of low-level shockwave exposure to the brain, we first sought to establish a level of blast that would produce no detectable evidence of lung barotrauma. Animals were placed on a foam bed and fitted with miniature noise reduction inserts to eliminate potential neurological injury due to loud sounds or tympanic rupture (Saljo et al., 2003). At all exposure levels (i.e., 11.5 kPa, 35 kPa, and 66 kPa), we observed no mortality or external signs of injury. Startle reflexes in response to snapping or clapping following shock-wave exposure was maintained, suggesting maintenance of hearing in shock-wave-exposed animals fitted with noise reduction inserts. At 11.5 kPa of overpressure exposure H&E lung pathology was virtually identical to that of naïve animals. Lung structure displayed no evidence of collapsed alveoli, hemorrhage due to trauma, or increased presence of resident macrophages (Fig. 2A and B). At 66 kPa overpressure there was notable evidence of lung hemorrhage and accumulation of inflammatory cells in the bronchiolar ducts, as well as fragmentation of the alveolar structure (Fig. 2C). Examination of BALF cell density was comparable between naïve and 11.5-kPa-exposed animals. Nuclear identification of cell types indicated that the vast majority were alveolar macrophages, as evidenced by the large single-nucleated morphology with some interspersed red blood cells in both naïve and 11.5-kPa-exposed animals (Fig. 2D and E). There was no evidence of increased neutrophil or granulocyte infiltration in BALF 24 h following 11.5-kPa shock-wave exposure. The number of alveolar macrophages was 154.3±7.0 cells in naïve animals and 157.0±8.5 cells in 11.5-kPa shock-wave-exposed rats (Fig. 2H). There was an increase in macrophage density at 35 kPa, but comparable red blood cell content, to naïve animals and those exposed to 11.5 kPa (Fig. 2F). By contrast, in 66-kPa-exposed animals we observed an obvious increase in the red blood content in the BALF (Fig. 2G). Macrophage density appeared lower, but was likely a result of clumping from the BALF spindown that we were unable to reconstitute, even with vigorous pipetting.
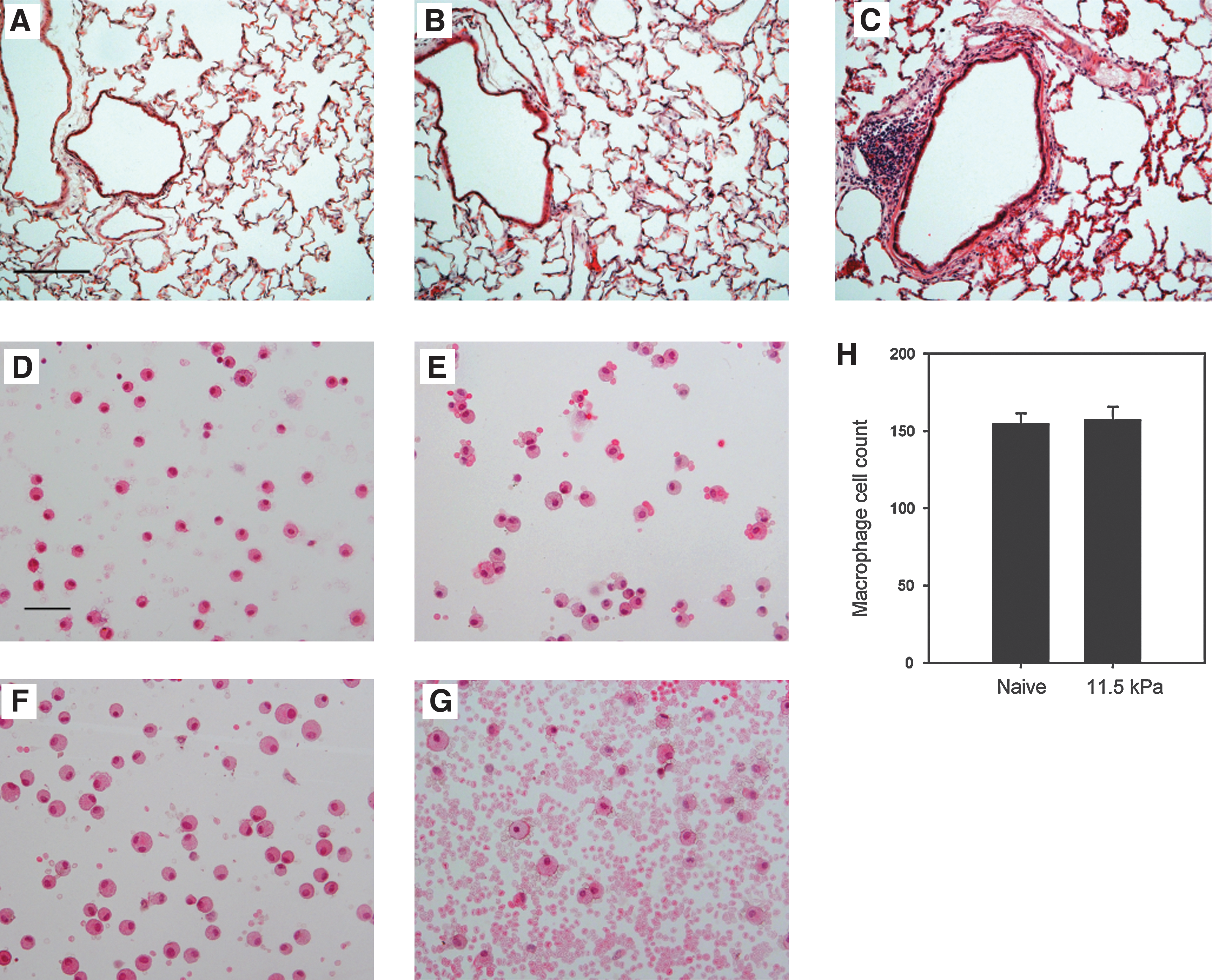
Representative hematoxylin and eosin (H&E) images of bronchiolar lavage (BAL) lungs from naïve (
Regionalized TUNEL distribution following low-level shock-wave exposure
Having established a level of sub-pulmonary barotrauma shock-waves, we next assessed for evidence of cell death using TUNEL to identify vulnerable brain structures. Quantification of TUNEL-positive cells was done for the cortex, hippocampus, corpus callosum, and periventricular region. Based on the effects of shockwaves traversing media of differing densities as described by theoretical models such as Richtmyer-Meshkov instabilities (Richtmyer, 1960), fluid dynamics experiments (Brouilette, 2002), and biological systems (Guy et al., 1998), we hypothesized that brain regions with the greatest differentials in tissue density, such as fluid-tissue interfaces (e.g., the periventricular region), or regions delineating grey and white matter separations, would be most susceptible to shock-wave-induced trauma. TUNEL counts in the cortex and hippocampus were comparable for naïve and shock-wave-exposed animals, with fewer than 1 TUNEL-positive cell detected per square millimeter. In the corpus callosum the number of TUNEL-positive cells was significantly different between treatment groups by one-way ANOVA (p=0.04; Fig. 3A–C). Post-hoc analysis demonstrated a significant difference between the naïve and shock-wave-exposed groups (p=0.05 at 24 h and p=0.03 at 72 h relative to naïve animals). The periventricular region demonstrated the greatest density of TUNEL distribution per unit area relative to other brain regions. Values between naïve, 24 h, and 72 h post-shock-wave-exposed groups were significantly different (p<0.001). Post-hoc analysis indicated a significantly greater number of TUNEL-positive cells per unit area at 24 and 72 h post-blast relative to naïve animals (p<0.001 for both time points). There was also a significantly greater density of TUNEL-positive cells at 72 h than at 24 h post-injury in the periventricular region (p=0.05). In order to confirm that the TUNEL distribution was not a result of differences in overall cell density in the assessed regions, we quantified the total number of cells in the assessed regions. The average number of nuclei per square millimeter of brain region used in the TUNEL analysis was 2771±101 in the cortex, 2823±501 nuclei per mm2 in the hippocampus, 3215±129 in the corpus callosum and 6764±329 in the periventricular region. The ratio of nuclei distribution for these regions using the cortex as a baseline value was approximately 1:1, 1:1.02, 1:1.16, and 1:2.44, for the cortex, hippocampus, corpus callosum, and periventricular regions, respectively. Although there was a higher density of cells in the corpus callosum and periventricular region, this did not account for the disproportionately larger increase in TUNEL values in these regions. Moreover, the nuclei sampling in the hippocampus was based on an average of sampling areas from the CA1, dentate gyrus, and regions with lower density between. The dentate gyrus and CA1 regions alone had cellular densities of 11,677±1691 nuclei/mm2, which was 4.2 times greater than the density seen in the cortex. Given the relative absence of TUNEL expression in the hippocampus and cortex, in conjunction with the assessment of overall cell density, the results suggest that the distribution of TUNEL-positive cells was not due to skewing from cellular density differences in these regions.
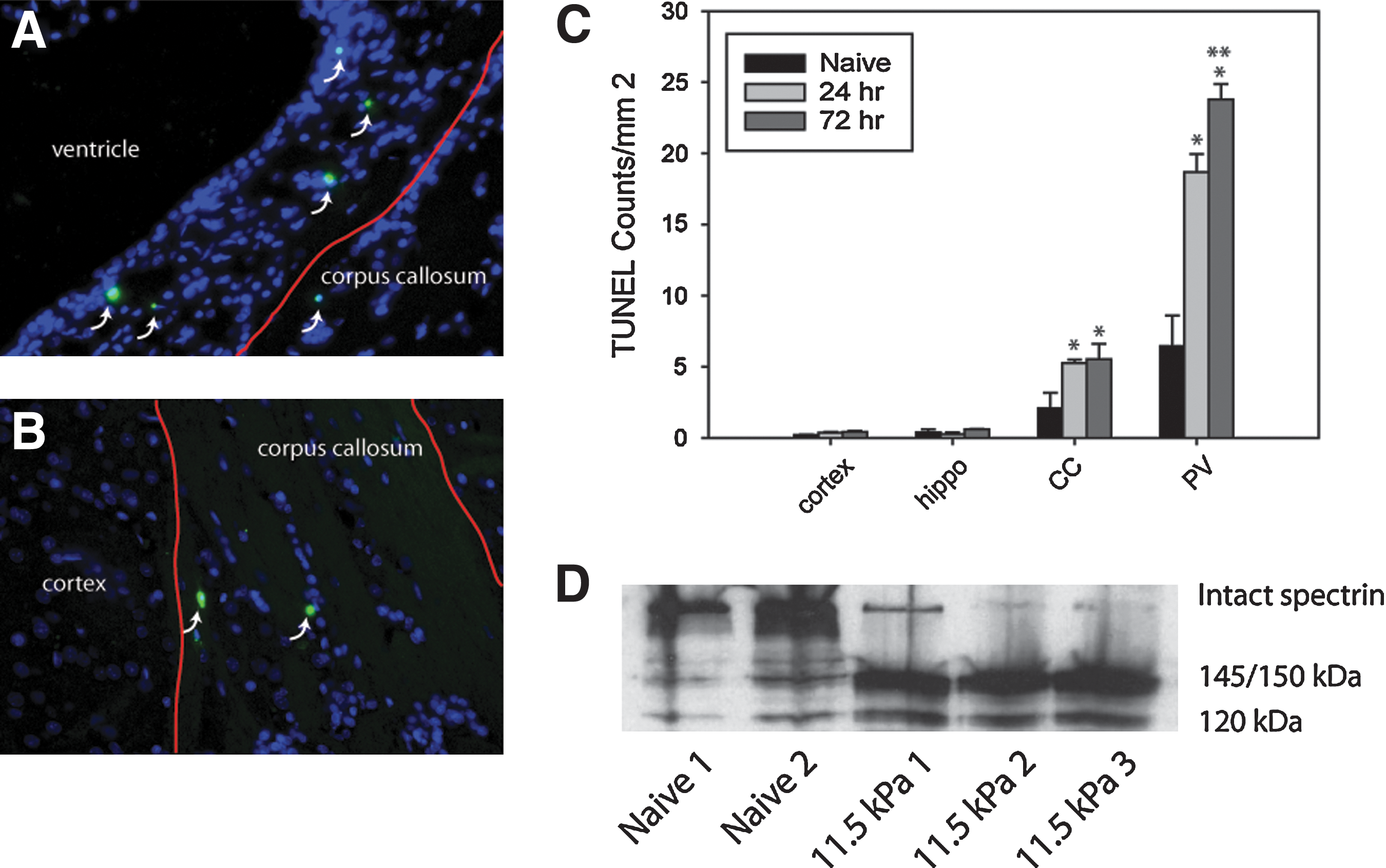
(
Since DNA fragmentation can also occur in necrotic forms of cell death (Grasl-Kraupp et al., 1995), we examined shock-wave-exposed rat tissue for expression of the caspase-3-mediated 120-kDa breakdown fragment of spectrin (data for hippocampus samples are shown in Fig. 3D). The results suggest that the TUNEL-positive cells we observed may have been undergoing apoptosis. However, this does not rule out the possibility that the observed cell death was also in part caused by secondary necrotic phenomena. As a whole, the total relative amount of detectable cell death was minimal, and was primarily used as a means to detect potentially vulnerable brain structures. In our model the pattern suggested vulnerability in the vicinity of a major white matter tract or periventricular region. Given these findings we then examined the expression of various white matter–associated cytoskeletal proteins.
Altered expression of axonal cytoskeletal proteins, αII-spectrin, and NF200 following shock-wave exposure
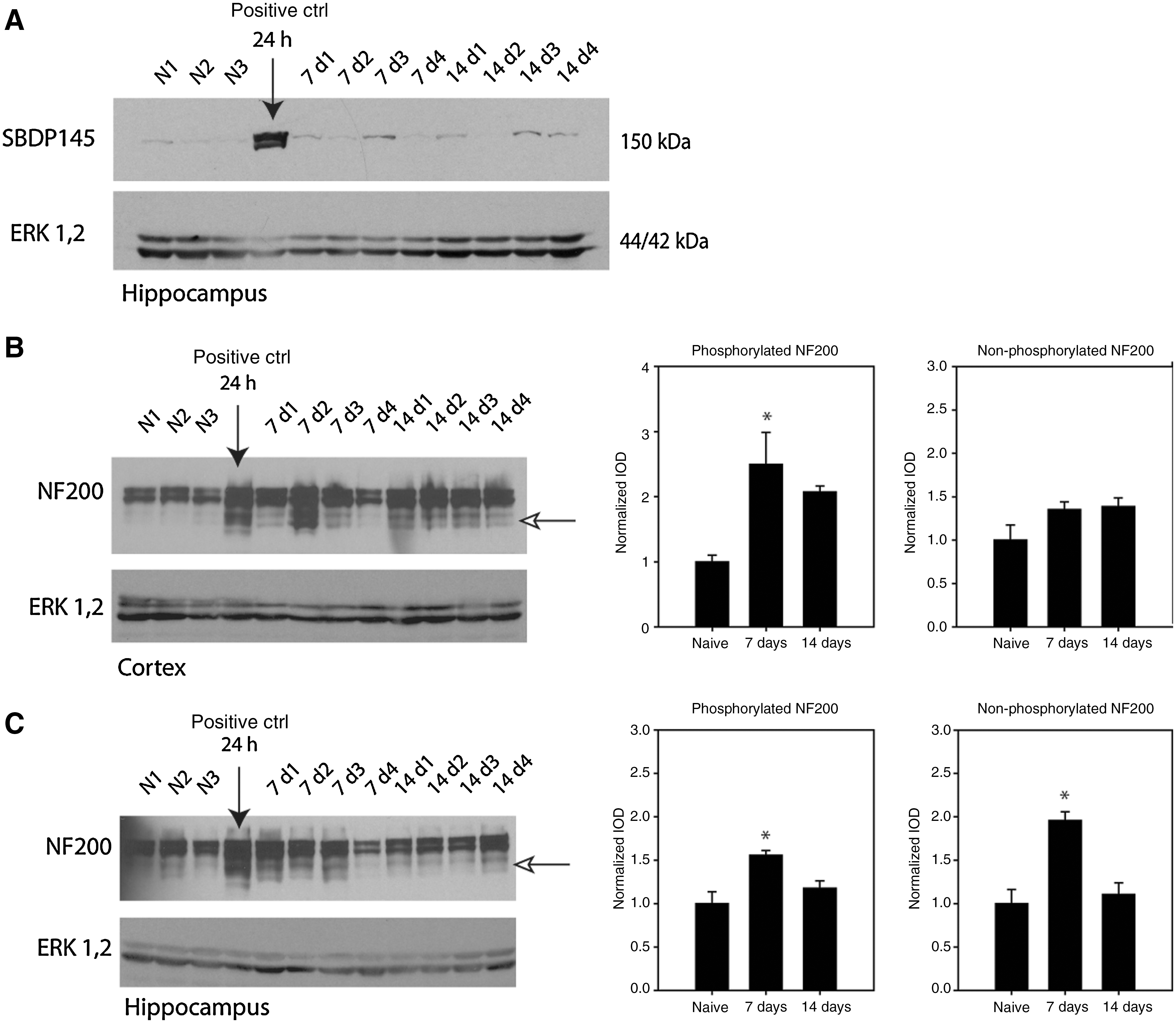
Given the relatively high distribution of TUNEL-positive cells in the corpus callosum compared to the cortex and hippocampus, we examined the expression of a predominantly axonally-expressed cytoskeletal protein, αII-spectrin, which is known to be vulnerable to calpain-mediated proteolysis in models of traumatic brain injury (Buki and Povlishock, 2006; Park et al., 2008). A time-course examination of calpain-mediated degradation of αII-spectrin indicated that proteolysis occurred at 12 h post-injury, but not before 6 h post-injury (data not shown for the 3-h time point), in both the cortex and hippocampus (Fig. 4A and B). Calpain-mediated spectrin breakdown products (SBDP) were detected up to 72 h post-injury (Fig. 4C), and relatively low-levels were seen at 7 and 14 days (Fig. 5A).
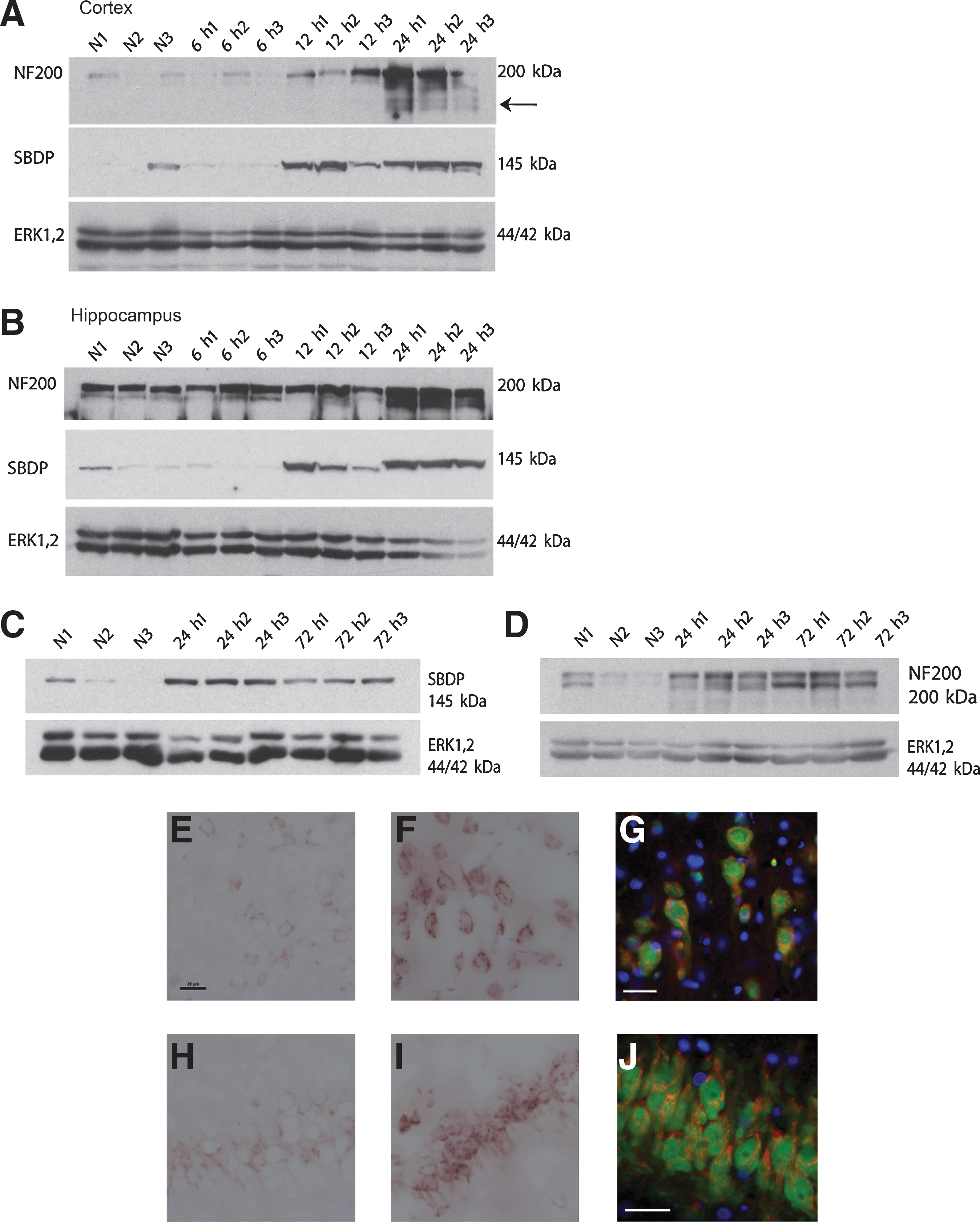
(
(
An assessment of the localization of spectrin breakdown by immunoperoxidase staining indicated a diffuse distribution in the cortex, while naïve animals had minimally detectable levels of expression (Fig. 4E and F). Similar expression was observed in the CA1 hippocampus region at 24 h post-injury (Fig. 4H and I). Spectrin breakdown was localized primarily to the cell bodies; however, there was evidence of labeling within the proximal axon branch as well. Double-label immunofluorescence with NeuN confirmed that cells expressing spectrin breakdown fragments were neurons (Fig. 4G and J).
We examined the expression of the heavy neurofilament protein NF200, also an abundantly expressed protein in myelinated axons. In the acute phase of injury from 6 to 24 h, there was a delayed but notable difference in the response of NF200 to low-level shock-wave exposure. In the cortex there were low-levels of NF200 expression in naïve animals, comprised predominantly of the phosphorylated isoform (i.e., the upper band of the NF200 doublet). By 12 h post-injury there was an increase in NF200 expression. This increased expression became more pronounced at 24 h post-injury, and was accompanied by the presence of a lower- molecular-weight breakdown fragment (Fig. 4A). Similarly, in the hippocampus the NF200 doublet displayed a greater degree of phosphorylated neurofilament expression in naïve and 6-h-injured tissues. There was a transient loss of the non-phosphorylated band at 12 h, with a subsequent increase in both forms of NF200 at 24 h post-injury (Fig. 4B). The increase in heavy neurofilament expression was also observed at 72 h (Fig. 4D). Despite the observed decrease in calpain-mediated SBDP at 7 and 14 days post-injury, phosphorylated NF200 expression at these time points was significantly different from naïve levels in the cortex (p=0.001), and hippocampus (p=0.016; Fig. 5B and C). Post-hoc analysis indicated that values were significantly elevated at 7 days post-injury in the cortex and hippocampus (p<0.05), relative to naïve levels. The expression of non-phosphorylated NF200 was not significantly different in the cortex at 7 and 14 days post-injury relative to naïve levels. By contrast, in the hippocampus, the expression of non-phosphorylated NF200 was significantly increased relative to naïve animals at 7 days post-injury (p<0.05). There was also the presence of NF200 breakdown products seen at 7 and 14 days post-injury in both the cortex and hippocampus (indicated by the open arrows in Fig. 5B and C). Note that the representative blots have been overexposed to illustrate the presence of NF200 breakdown products, and were not used in the quantitative analysis of NF200 expression.
In summary, early spectrin breakdown is paralleled by a pathophysiological increase in heavy neurofilament expression at concurrent post-injury time points. At chronic time points, at which spectrin degradation was minimal, there was a persistent increase in NF200 expression at 7 days post-injury. These results suggest that the cytoskeletal response to shock-wave trauma is delayed and heterogeneous, dependent on the type of affected cytoskeletal protein.
Expression of CNPase and electrophysiological function following low-level shock-wave exposure
The susceptibility of white matter function to low-level shock-wave exposure was evaluated in the corpus callosum through evoked CAP recordings. Similarly to our previously described methods (Baker et al., 2002), evoked CAPs from the corpus callosum in acute slice preparations were evaluated in naïve animals, and 1-, 3-, 7-, 14-, and 30-day post-injury rats (Fig. 6A). An in/out curve was generated to evaluate the maximum evoked CAP response at 1.5-mA stimulation. Mean CAP values in the myelinated fibers were 2.59±0.18 mV in naïve rats, 2.34±0.24 mV at 1 day, 1.8±0.16 mV at 3 days, 2.49±0.15 mV at 7 days, 2.54±0.25 mV at 14 days, and 2.36±0.17 mV at 30 days post-injury (Fig. 6B). Although the CAP values were not significantly different (p=0.08), the largest decline in mean CAP value correlated with the decrease in CNPase expression observed at 24 h and 3 days post-injury (Fig. 6F–H). In unmyelinated fibers there was a significant difference between mean CAP values (p=0.01). Mean CAP values were 1.58±0.15 mV in naïve rats, 1.58±0.23 mV at 1 day, 0.97±0.10 mV at 3 days, 1.10±0.08 mV at 7 days, 1.33±0.16 mV at 14 days, and 1.04±0.12 mV at 30 days post-injury. Post-hoc analysis indicated that a significant decline in CAP amplitude was seen at 3 days (p=0.05) relative to naïve CAP levels (Fig. 6C). In contrast to myelinated CAP values, which largely recovered by 7 days post-injury, unmyelinated CAP amplitudes remained depressed at 30 days post-injury.

(
Duration of the CAP amplitudes was significantly different in myelinated fibers across post-injury time points and in naïve animals (p=0.05). Mean CAP duration values were 2.17±0.10 msec in naïve rats, 2.20±0.08 msec at 1 day, 2.11±0.08 msec at 3 days, 2.30±0.11 msec at 7 days, 2.51±0.09 msec at 14 days, and 2.48±0.16 msec at 30 days post-injury (Fig. 6D). Post-hoc analysis indicated that values at 14 and 30 days post-injury were significantly longer than CAP duration in naïve rats (p=0.05). The difference in mean CAP durations with unmyelinated fibers approached significance (p=0.07). Mean values in the study groups were 2.46±0.09 msec in naïve rats, 2.59±0.09 msec at 1 day, 2.48±0.11 msec at 3 days, 2.65±0.12 msec at 7 days, 2.90±0.09 msec at 14 days, and 2.78±0.18 msec at 30 days post-injury (Fig. 6E). Similarly to the pattern of CAP duration change in myelinated fibers, the unmyelinated CAP duration generally increased at 14 and 30 days post-injury.
CNPase expression changed significantly at 24 h and 3 days post-injury in the hippocampus (p=0.03) and cortex (p=0.02; Fig. 6F–H). Post-hoc analysis indicated a significant decline in CNPase expression in both regions of the brain at these time points relative to naïve levels of expression (p<0.05). Collectively, the electrophysiological results and the expression of the myelin-associated protein CNPase provide further evidence of white matter pathophysiological involvement in the response to low-level shock-wave exposure.
Discussion
Blast injuries have been categorized as a novel mechanism of injury to the brain, with overlapping yet distinct clinical features that differ from those of conventional closed and penetrating brain traumas (Cernak and Noble-Haeusslein, 2009; Ling et al., 2009). While the majority of mild traumatic brain injury (mTBI) cases resolve over time, a significant proportion go on to develop persistent neurological and psychological impairments (Hoge et al., 2008). The cause of mTBI and post-traumatic stress disorder (PTSD) symptoms in the battlefield is highly controversial (Vasterling et al., 2009). However, there is some evidence to suggest that exposure to explosions results in a greater likelihood of neurological deficits and impairments (Ruff, 2008). Given the overlap in clinical symptoms of mild TBI and PTSD (Stein and McAllister, 2009), the etiology of these disease states stemming from experiences in a life-threatening environment implicates either physical blast trauma, occupational stresses, or both, as causative factors contributing to neurological and psychological impairments (Hoge et al., 2008; Mora et al., 2009).
Lethality curves, generated by Richmond's study on blast loading in various mammalian species, suggest that amplitude and duration are critical factors in determining thresholds for mortality (Richmond et al., 1968). While this study clearly demonstrated a subject-mass dependency following blast exposure, the primary cause of death in these high-overpressure blast experiments was presumably lung trauma or air embolism. Furthermore, Bowen's scaling calculations for small animals was also based on injury mechanisms in the lungs related to mass-spring oscillating systems (Bowen et al., 1968). To date, there is no standardized scale that describes the influence of blast wave parameters (i.e., overpressure, duration, and frequency) as a function of subject mass as it relates to brain injury. Variables such as bone porosity, calcium content, bending strength and Young's modulus of elasticity, will vary from species to species (Currey, 1999). Assuming an interaction with the skull in the transmission of a shock-wave impulse to the brain (Moss et al., 2009), the classification of varying ranges of overpressures to rodents as mild, moderate, or severe blast-induced TBI is unknown. As such a scaling metric for blast brain injuries that can be applied to specific species remains to be elucidated. Therefore we sought to investigate a direct causal mechanism of shock-wave-induced brain trauma in a low-intensity model in which lung injury was not present. Since the mechanisms of shock-wave transmission across the skull and brain are not yet fully understood, we are unable to create a scale that takes into account mechanisms of shock-wave transmission in rats that could be extended for use in larger animals, such as those described by Richmond and associates (Richmond et al., 1968). However, our overpressure amplitude and durations, which were notably lower than those seen in previous reports in rodent models, were adjusted to a level that produced little or no discernible lung trauma, but that at higher levels could induce lung barotrauma. This suggests a scaling factor that takes into account the decreased body mass and skull thickness of rodents by lowering the overall impulse of the shockwave (i.e., amplitude and duration), rather than subjecting them to the equivalent pressure amplitudes and durations used in large mammal studies.
Champion and colleagues described the disconnect between laboratory studies of primary blast and injuries that occur in the field, suggesting that many studies may be irrelevant to real-world conditions (Champion et al., 2009). For example, in a recent review of primary blast studies, pulmonary barotraumas were absent when animals were more than 20 m from a 100-kg TNT explosion (Bass et al., 2008). Champion's group contends that at any closer distance, there would be significant injury or death associated with secondary blast injury components, thus the isolation of specific effects on the brain of shockwaves is limited in interpretation, as this would not include the full spectrum of blast trauma. In the current study we sought to investigate biological mechanisms that were uncomplicated by systemic injuries related to secondary or tertiary blast injuries.
The adjustment of the shockwave in our system to a level that resulted in virtually no lung injury likely accounts for the differences in our observed effects on the brain with previously reported findings. The distribution of TUNEL in regions with different densities, such as that observed in the periventricular region, was in accordance with the characteristics of shock-wave behavior described by Richtmyer-Meshkov instabilities, whereby disturbances are created at the interface of fluids impulsively accelerated by a shockwave (Meshkov, 1969). Recent mathematical modeling of low-amplitude shockwaves traversing anisotropic soft tissues also suggests that the orientation of the white matter fiber tracts in the corpus callosum may play a role in susceptibility to shock-wave-mediated injury (Namani and Bayly, 2009), which may partially explain our observed increase in TUNEL-expressing cells in this region relative to the cortex and hippocampus. The relatively low distribution of TUNEL-positive cells in our blast model was consistent with a model of mTBI in which the primary contributors to neuropathology are likely related to cell dysfunction rather than cell death. The significantly higher number of TUNEL-positive cells in the periventricular regions in comparison to the corpus callosum, hippocampus, and cortex, further supports our hypothesis of density-shearing effects due to shockwaves, given the larger density differential between CSF and brain tissue than that between grey and white matter. Interestingly, the cortex and hippocampus demonstrated a diffuse distribution of calpain-mediated SBDP, indicating that some pathological cytoskeletal alterations occurred in these regions as well. Despite the diffuse distribution of SBDP, it is unlikely that these neurons were destined for death, given the comparatively low number of TUNEL-positive cells quantified. Under high-amplitude and long-duration shock-wave conditions, these non-lethal cellular alterations could conceivably be masked by overt cell injury and death throughout the brain (Long et al., 2009). Given the temporal range of SBDP expression up to 3 days post-injury, it is possible that the breakdown of spectrin represents a transient remodeling of cytoarchitecture in response to perturbations caused by a shock-wave impulse. In this scenario, synaptic or cytoskeletal remodeling may be a more appropriate representation of the altered neurological function associated with mTBI, as opposed to extensive cell loss. A critical issue that remains to be investigated is whether this period of cytoskeletal remodeling represents a period of increased neurological vulnerability to injury from repeated exposure to shockwaves, or if this remodeling results in permanent pathophysiological changes in brain function.
There was evidence to suggest white matter involvement in the brain's response to low-level shock-wave exposure, as indicated by electrophysiological data and CNPase expression. Although we were unable to isolate white matter tissue in our Western blot tissue preparations, there was evidence of cytoskeletal proteolysis of NF200 and spectrin, proteins known to be expressed in high abundance in axons. The increased expression of NF200 may be related to accumulation of neurofilaments in the neuronal somata, as was previously reported in a recent model of primary blast trauma (Saljo et al., 2000). Interestingly, that group reported an accumulation of the phosphorylated form of NF200 in the neuronal perikarya. The significance of their findings may relate to heavy neurofilament dynamics and rates of axonal trafficking, which are closely linked to their phosphorylation state (Shea and Chan, 2008). Increased phosphorylation is associated with slower axonal transport rates due to reduced binding with the motor protein kinesin (Jung et al., 2005). An impairment in proper neurofilament subunit trafficking dynamics results in the accumulation of neurofilaments, and is a suspected cause of several neurodegenerative disorders, including amyotrophic lateral sclerosis, Alzheimer's disease, Charcot-Marie-Tooth disease, and Parkinson's disease (Liu et al., 2004; Rao and Nixon, 2003). Our results suggest a possible impairment of neurofilament transport similar to those observed in other types of neuropathologies.
Our electrophysiological results are also consistent with the hypothesis of impaired neurofilament transport contributing to white matter dysfunction. Heavy neurofilament assembly and phosphorylation regulate axonal caliber, thereby directly influencing the functional properties of the axon (Perrot et al., 2008; Sihag et al., 2007). The increased duration of the CAP response in shock-wave-exposed rats suggests impairment in axonal function, possibly related to a decrease in axonal caliber resulting in reduced action potential velocity. There was relatively strong agreement between the temporal expression of NF200, which displayed an increased level of expression relative to naïve animals up to 14 days post-injury, and the detection of increased CAP durations at this chronic time point. Whether the observed presence of NF200 breakdown fragments at 7 and 14 days post-injury is also involved in axonal dysfunction is unclear. It is possible that the breakdown of NF200 is in response to excessive accumulation in the neuronal somata due to impaired axonal trafficking; however, this has yet to be validated.
The decrease in CNPase expression seen at 1 and 3 days post-injury was also in agreement with our electrophysiologic data, which indicated a transient decrease in myelinated CAP amplitude at 3 days post-injury. At 7 and 14 days, the levels of CNPase expression recovered to levels comparable to those seen in naïve animals (data not shown), suggesting a possible transient decoupling of the axon-myelin interaction. Ultrastructural electron microscopy studies will be required to further validate these results. It is clear, however, that the white matter response to shockwaves is complex, involving numerous pathophysiological mechanisms that evolve well beyond the initial exposure.
It is interesting to note the delayed temporal profile of the molecular and functional changes seen in our blast model. We observed delayed proteolysis of αII-spectrin, occurring at 12 h post-injury, and chronic deficits in CAP durations, seen only by 14 days post-injury. These data not only demonstrate a progressive and persistent pathophysiological response to low-level shockwaves, but may also have significant implications for potential windows of opportunity for the pharmacological treatment of shock-wave-mediated pathophysiology.
Despite the blast-induced molecular and functional deficits described in this study, the overall effects on behavioral function remain to be elucidated. Our shock-wave-exposed animals did not exhibit any obvious signs of abnormal behavior. However, a comprehensive evaluation of specific tasks involving memory, learning, or stress responses, following low-level shock-wave exposure remains to be performed. There is some evidence to suggest a possible impairment in memory, as Saljo and colleagues reported deficits in spatial learning in rats subjected to 10-kPa and 30-kPa overpressures at 2 days post-injury (Saljo et al., 2009b). Given the subtlety of the electrophysiological deficits, and the low numbers of TUNEL-positive cells seen in this study, it seems plausible that a collection of micro-impairments may contribute to overall behavioral deficits in certain domains of neurological function.
Conclusion
Non-fatal, low-level primary blast intracranial insult has been suspected as a potential cause of mild traumatic brain injury, the so-called signature injury of the Iraq War, but the etiology remains obscure and early diagnosis elusive. Our data demonstrate a complex series of cellular events occurring due to energy transfer to the brain associated with shock-wave exposure. The delayed nature of these changes suggests an opportunity for intervention, including not only prophylactic measures, but also post-blast exposure intervention. This otherwise occult brain injury due to shock-wave exposure resulted in a distinct pattern of delayed cell death and the development of chronic electrophysiological impairments. The further elucidation of shock-wave-induced brain trauma is critical for the development of effective post-exposure intervention strategies.
Footnotes
Acknowledgments
We thank David Ritzel of Dyn-FX Consulting for his technical input on primary blast modeling, and Elaine Liu for technical wet bench assistance.
This work is funded through the Public Works Government Services Contract, Department of Defense Research Canada, PWGSC contract #W7711-078105/A.
Author Disclosure Statement
No competing financial interests exist.