
Abstract
The temporal and regional expression profiles of matrix metalloproteinase-9 (MMP-9), after moderate or severe traumatic brain injury (TBI) were measured to investigate the effects of post-traumatic hypothermia (33°C) or hyperthermia (39°C). In the first phase of this study, adult male Sprague-Dawley rats were randomly assigned to groups of moderate TBI (1.8–2.2 atm), severe TBI (2.4–2.7 atm), and sham-injured control. The rats were killed at 4, 6, 12, 24, 48, and 72 h, or 1 week after TBI, for mRNA and protein analysis. In the second phase, rats underwent moderate fluid percussion brain injury, followed immediately by 4 h of post-traumatic normothermia (37°C), hyperthermia (39°C), or hypothermia (32°C). The rats were killed at 12 and 48 h after TBI for mRNA expression analyses, or killed at 24 and 72 h after TBI for protein expression analyses. Brain samples, including the cerebral cortex and hippocampus (both ipsilateral and contralateral hemispheres of each group), were assayed using RT-PCR and Western blot techniques. MMP-9 levels in both the ipsilateral and contralateral hemispheres were significantly increased after TBI compared with those of sham injured animals (p < 0.01). Two expression peaks of MMP-9 were observed in the ipsilateral cortex and hippocampus. An increase in injury severity was associated with an increase in mRNA (12 and 48 h), and protein (24 and 72 h) levels of MMP-9. Post-traumatic hypothermia attenuated the increase in both the mRNA and protein levels of MMP-9, compared with normothermia and hyperthermia (p < 0.01). In contrast, hyperthermia had no significant effect on mRNA (at 12 h) and protein levels (at 24 h) of MMP-9, compared with normothermic values (p > 0.05), but resulted in a significant increase in the levels of MMP-9 mRNA and protein at 24 and 72 h, respectively (p < 0.01). Increases in MMP-9 mRNA and protein after TBI were proportional to injury severity in this model. The effects of post-traumatic hypothermia on the expression of MMP-9 may partially explain the observed effects of post-traumatic temperature on secondary injury after TBI.
Introduction
Matrix metalloproteinases (MMPs) are a family of extracellular zinc- and calcium-dependent endopeptidases that degrade the components of extracellular matrix (ECM; Rosenberg, 2002;Wang and Tsirka, 2005). Specifically, MMP elevation was demonstrated after TBI (Noble et al., 2002; Wang et al., 2000), as well as after cerebral ischemia (Burk et al., 2008; Rosenberg et al., 1996). A role of MMPs, MMP-9 in particular, has been suggested in the pathogenesis of neurological disorders (Rosenberg et al., 1996; Yong et al., 2001). MMP-9 can cause an increase in capillary permeability and produce brain edema secondary to ischemia and TBI (Gasche et al., 1999; Rosenberg and Navratil, 1997). Thus MMP inhibition would be expected to attenuate secondary insults after injury. Consistent with this hypothesis is the finding that administration of the MMP inhibitors BB-1101 and BB-94 reduced brain edema and mortality following intracerebral hemorrhage induced by collagenase or tissue plasminogen activator (Rosenberg and Navratil, 1997). Treatment with a pan-MMP inhibitor was also reported to reduce MMP permeability and hemorrhage after ischemic reperfusion (Rosenberg et al., 1997; Sumii and Lo, 2002). However, absolute MMP-9 inhibition in the form of knockout mice has yielded conflicting results with regard to pathological and neurological outcomes (Tang et al., 2004). A more complete understanding of the injury-induced MMP-9 response, as well as the temporal profile of biochemical alterations, is crucial to resolve these controversies and to design effective therapeutic interventions.
Several clinical works and experimental studies using a model of TBI have shown the neuroprotective effects of hypothermia, including inhibition of neurological injury (Jiang et al., 2000; Marion and White, 1997; Polderman et al., 2002), reduction in infarct size (Celik et al., 2006; Resnick et al., 1994), and improvement in neurological outcome (Bramlett et al., 1995). However, whether hypothermia influences the expression of MMP-9 after injury remains to be elucidated. The purpose of this study was thus to investigate whether TBI severity affects the temporal and regional expression profiles of MMP-9, and also to determine the effects of post-traumatic temperature manipulations.
Methods
Animals
The protocol for these animal studies was approved by the Animal Care and Experiment Committee of the School of Medicine, Shanghai JiaoTong University. The experiments were performed with male Sprague-Dawley rats weighing 320−380 g (n = 250). All animals were maintained for at least 7 days before the study in an air-conditioned room with a constant temperature (23–25°C), with a 12-h light/dark cycle. The rats were allowed free access to water, but food was withheld overnight before surgery.
Experimental groups
The rats were divided into three groups based on injury severity (Table 1). Group 1 included sham-injured rats subjected to all surgical procedures except TBI. Group 2 was the moderate TBI group, in which injury was produced using fluid percussion pressures of 1.8−2.2 atm. Group 3 was the severe TBI group, in which injury was produced using fluid percussion pressures of 2.4−2.7 atm.
RT-PCR, reverse transcriptase polymerase chain reaction; TBI, traumatic brain injury.
In the first phase of the study the regional and temporal profiles of MMP-9 mRNA (n = 5/group) and protein (n = 5/group) levels in brain tissue were determined after moderate or severe fluid percussion brain injury. It was reported by other researchers that pathophysiological changes were noted at 1 h, and peaked at 24 h, after TBI due to impact or penetration. Thus we used 4, 6, 12, 24, 48, and 72 h as the acute time points to be analyzed temporally (Kinoshita et al., 2002; Vitarbo et al., 2004). To establish temporal profiles for MMP-9 mRNA and protein expression, brain tissue samples were assayed 4, 6, 12, 24, 48, and 72 h and 1 week after moderate fluid percussion injury or severe TBI.
In the next phase of the study, the systemic post-traumatic brain temperature was manipulated to determine its effects on MMP-9 mRNA and protein levels. For this study, temperature manipulations were conducted for 4 h immediately after TBI. The animals were divided into four groups as follows: 32°C after moderate TBI, 37°C after moderate TBI, 39°C after moderate TBI, and 39°C after sham injury. To achieve the target temperatures, the rats were placed in a small plastic container and were kept hyperthermic (39°C) or hypothermic (32°C) by using a heating lamp or cooled air, respectively, beginning immediately after TBI. Some of the rats were killed 12 and 48 h after TBI for analysis of mRNA expression. Other groups were killed 24 and 72 h after TBI for analysis of protein expression.
Surgical preparation
The animals were anesthetized with a nitrous oxide/oxygen mixture (70%/30%) containing 2% halothane. Tracheal intubation was performed, and positive pressure ventilation was initiated. Both the femoral artery and vein on the right side were cannulated with polyethylene tubing for monitoring of blood pressure and blood gas analysis. After cannulation the wound was sutured, and the animals were turned to the prone position. The animals were then placed in a stereotactic frame and the scalp was incised sagittally. A 4.8-mm-diameter skull trephine opening was drilled 2 mm to the left of the midline between the bregma and the lambda. Two fixation screws were then placed 1 mm rostral to the bregma and 1 mm caudal to the lambda. A Luer-Lok (Becton Dickinson, Mountain View, CA) needle hub was placed in the skull hole and cemented in position with cyanoacrylate glue. Dental acrylic was then poured around the needle hub and the two screws for rigidity. A 1.5-mm burr hole was then drilled at the middle of the occipital bone 1.5 mm from the midline and 1.5 mm from the horizontal edge. Dental acrylic was used to immobilize the probe. A guide (a modified 20-gauge 1.5-in. needle) for the brain temperature probe was fixed to the surface of the skull over the burr hole with cyanoacrylate glue and dental acrylic. During the entire experiment the mean arterial pressure was monitored continuously, and blood gases were measured before injury and 15 min after injury.
Fluid percussion traumatic brain injury
A fluid percussion device was used to produce TBI as described previously (Dixon et al., 1987). Briefly, the device consists of an acrylic glass cylindrical reservoir 60 cm long and 4.5 cm in diameter, with one end having a rubber-covered acrylic glass piston mounted on O rings. The opposite end is fitted with a transducer housing ending with a 2.6-mm inside diameter Luer-Lok fitting. The entire system is filled with 37°C isotonic saline. Injury is induced by the descent of a metal pendulum striking the piston, thereby injecting a small volume of saline epidurally into the closed cranial cavity, producing a brief displacement and deformation of the neuronal tissue. The resulting pressure pulse is measured in atmospheres by an extracranial transducer (Statham PA 85-100; Gould, Oxnard, CA) and recorded on a storage oscilloscope (Tektronix 5111; Tektronix, Beaverton, OR). The animals sustained a moderate (1.8−2.2 atm) or severe (2.4−2.7 atm) magnitude of injury, as previously described (Dixon et al., 1987).
Temperature manipulations
Frontal cortex brain temperature was monitored with a digital electronic thermometer (model DP 80; Omega Engineering, Inc., Stamford, CT) with a 0.15-mm-diameter temperature probe (model HYP-033-1-T-G-60-SMP-M; Omega Engineering) inserted 4.0 mm ventral to the surface of the skull. The probe was removed before fluid percussion injury and replaced immediately after injury. Rectal temperatures were measured with an electronic thermometer (model 43 TE; YSI, Inc., Yellow Springs, OH) and temperature probe (series 400; YSI). The brain temperature was 37°C before the hypothermic treatment. Hypothermia was achieved as described in a previous study (Jiang et al., 1991). Briefly, a brain temperature of 32°C was achieved by immersing the body of the anesthetized rat in an ice-cold water bath (0°C). The skin and fur of the animals were protected from direct contact with the water by placing the animal in a plastic bag (with the head exposed) before immersion in the water bath. The animals were removed from the water bath when the brain temperature had fallen to within 2°C of the target. It took approximately 15 min to reach the target brain temperature, which was maintained for 4 h under general anesthesia at room temperature by intermittent application of ice packs when needed. Gradual rewarming to normothermic levels (37°C) over a 90-min period as described previously (Koizumi and Povlishock., 1998) was used to avoid rapid rewarming that may have influenced secondary injury processes. A brain temperature of 37°C or 39°C was achieved under general anesthesia with a heating blanket for 4 h.
Reverse transcriptase polymerase chain reaction
Analysis of the time course of mRNA for MMP-9 was done on injured rats at 4, 6, 12, 24, 48, and 72 h and 1 week. Five rats were studied at each time point for comparison with uninjured animals. Tissue from the cerebral cortex and hippocampus was homogenized in Trizol reagent (Invitrogen, Carlsbad, CA) for extraction of total RNA according to the manufacturer's protocol. Total RNA was quantitated using the 8453 UV-visible spectroscopy system (Agilent Corporation, Palo Alto, CA). First-strand cDNA was generated from total RNA (2 μg) according to the manufacturer's instructions for the reverse transcription system (Promega Corp., Madison, WI). The MMP-9 mRNA levels in each sample were determined by real-time PCR using SYBR® Green I Dye. One cDNA sample was diluted (gradient 1:10, 1:50, 1:100, 1:500, and 1:1000) as standard for real-time PCR on SYBR green-based detection. The reverse transcription product was included in the SYBR Premix Ex Taq™ kit (Perfect Real Time; TaKaRa, Ltd., Shiga, Japan), along with rat MMP-9 primers, and the mixture was placed in a real-time PCR thermal cycler (Lightcycler System; Roche Diagnostics Corp., Indianapolis, IN). Thermal cycling parameters were 30 sec at 95°C, followed by 40 cycles of 5 sec at 95°C, 5 sec at 60°C, and 30 sec at 72°C. At the end of the program, melting curve analysis was performed at 72°C for 30 sec, followed by a cooling step at 37°C for 30 sec. Each sample was also run with primers for a housekeeping gene. The following primer pairs were used: rat MMP-9, 5'-AGAAGGTGGATCCCCAGAG-3' and 5'- GGCCTTGTCTTGGTAGTGAAAG-3', and expression of the GAPDH gene, 5'-CCCCACACACATGCACTTACC-3', and 5'-CCTACTCCCAGGGCTTTGATT-3'. The relative expression levels for each gene of interest were calculated by normalizing the quantified cDNA transcript level (cycle threshold, Ct) to that of GAPDH. The data for MMP-9 RT-PCR represents values normalized to GAPDH, while GAPDH was maintained as a relative invariant throughout the study (Aswal et al., 2008; Jiang et al., 2009; Thellin et al., 1999).
Western blotting
Western blots were used to measure active MMP-9 (78 kD) protein of each animal in each group at all prescribed time points (the same as those for RT-PCR) after TBI. Brain tissue was homogenized by disrupting through syringes, and was then extracted in protein extraction buffer (1 M Tris-HCl [pH 6.8] 0.625 mL, 10% SDS 2 mL, and 50% glycerol 2 mL, with water added to make 10.00 mL). Total protein concentration was determined using the BCA protein assay kit (Bio-Rad Laboratories, Hercules, CA). Then 0.01% bromophenol blue and 10%β-mercaptoethanol were added to the protein sample, which was then boiled for 5 min. Total protein of 30 μg was separated on a 12−15% SDS-polyacrylamide gel by electrophoresis. The proteins were transferred to PVDF immunoblotting membranes (Bio-Rad Laboratories) and run at 120 volts for 1 h at 4°C. The membrane was blocked with 5% non-fat milk in phosphate-buffered saline (PBS) with 0.1% Tween-20, rinsed, and then incubated overnight at 4°C with the rabbit anti-MMP-9 antibody(1:100; Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-human β-actin (1:5000; Abcam, Cambridge, MA). After rinsing, the membranes were incubated with secondary antibody IRDye 800CW goat anti-rabbit fluorescently-labeled secondary antibody (1:2000; LI-COR Biosciences, Lincoln, NE), and IRDye 800CW goat anti-mouse fluorescently-labeled secondary antibody (1:5000; LI-COR Biosciences). The MMP-9 protein was detected immunologically using the LI-COR imaging system, which allows the detection of sub-picogram quantities of protein. After a 3-min incubation in the detection reagent, the blot was immediately exposed to Kodak diagnostic film (Eastman Kodak Co., Rochester, NY). Molecular weight and MMP-9 standard were included on each blot. The standards for MMP-9 were a kind gift of P. Alexander (Triple Point Biologics, Portland, OR). Gels from the independent hemispheres from all animals at each time point were run simultaneously to allow for comparisons between the different groups. The gels were scanned with a transparent scanner (Agfa DuoScan; Agfa Corp., Ridgefield Park, NJ), and the scanned images were quantified with a gel electrophoresis analysis program (Alpha Ease; Alpha Innotech Corp., San Leandro, CA). The relative levels of protein expression were calculated by normalizing to that of β-actin.
Immunofluorescence
Immunofluorescence was carried out on free-floating sections that were permeabilized for 1 h in 0.1% Triton X-100 buffer to reduce non-specific immunolabeling. Double immunostaining was performed by co-incubation of primary antibody overnight at 4°C. The primary antibodies and dilutions used were anti-MMP-9 (1:25; Alomone Labs, Jerusalem, Israel), anti-NeuN (1:1000 NeuN, neuronal nuclei-specific antibody; Chemicon International, Temecula, CA), and anti-GFAP (1:1000 glial fibrillary acidic protein [GFAP]; Chemicon). After rinsing, sample sections were incubated with Cy3-conjugated and Cy2-conjugated anti-mouse IgGs (both 1:500; Jackson ImmunoResearch West Grove, PA). The sections were mounted in Mowiol medium 4-88 (Calbiochem Corp., La Jolla, CA), and observed with a Bio-Rad MRC 1024 laser scanning confocal microscope equipped with a w40 oil-immersion objective. The images were exported into Adobe Photoshop 9.0 (Adobe Systems, San Jose, CA) for final processing. Staining without primary antibody served as negative controls.
Statistical analysis
Data are presented as mean ± SEM. A one-way analysis of variance (ANOVA) was used to compare the physiological variables between groups. Between-group comparisons of MMP-9 data were conducted using a two-way ANOVA. Post-hoc comparisons were made using Tukey's test. Differences were considered significant at p < 0.05.
Results
Physiological data
Physiological parameters were monitored for each group 15 min before and 15 min after TBI or sham procedures (Table 2). All physiological variables with the exception of temperature were within normal ranges for the TBI normothermic, TBI hyperthermic, and TBI hypothermic groups, as well as the sham group (p < 0.01).
PCO2, partial arterial carbon dioxide pressure; Po2, partial arterial oxygen pressure; MAP, mean arterial pressure; mTBI, moderate traumatic brain injury; sTBI, severe traumatic brain injury; TBI, traumatic brain injury.
Temporal and regional expression profiles of MMP-9 mRNA after moderate traumatic brain injury
The variation in the expression of MMP-9 mRNA in the ipsilateral and contralateral hemispheres over time after TBI is shown in Figure 1. The MMP-9 mRNA, as measured by quantitative RT-PCR, remained at a constant level over the tested time period in the sham group as shown in Figure 1 (the data for MMP-9 RT-PCR represent values normalized to GAPDH, and GAPDH was maintained as a relative invariant throughout the study; Aswal et al., 2008; Jiang et al., 2009; Thellin et al., 1999). At 4 h after TBI, the amount of MMP-9 mRNA in the ipsilateral and contralateral cortex and hippocampus in the severe TBI group increased significantly compared with the corresponding values in the sham group (p < 0.01). It kept increasing with time after TBI, and reached its maximal values around 12 h post-injury (in the ipsilateral cortex and hippocampus), and 48 h (in the contralateral cortex and hippocampus). A smaller peak in the amount of MMP-9 mRNA was observed at 48 h post-TBI on the ipsilateral side. MMP-9 mRNA expression in the moderate TBI group was also increased compared with that in the sham group at 4 h after TBI (p < 0.01). It also kept increasing in a similar trend as that of the severe TBI group, with the maximum values reached at the same time points in the cortex and hippocampus of both sides. However, this increase was significantly attenuated in this group compared with the severe TBI group. The maximum mRNA levels of MMP-9 in the moderate TBI group were significantly reduced, to 65.19 ± 2.40%, 71.52 ± 2.20%, 60.49 ± 2.30%, and 63.66 ± 1.80% of the corresponding values seen in the severe TBI group in the injured (ipsilateral) and uninjured (contralateral) hemispheres in the cortex and hippocampus, respectively. However, compared with the sham group, MMP-9 mRNA expression of the injured hemisphere in the moderate TBI group was still significantly higher at all time points except for 1 week (p < 0.01). At 1 week after TBI, the MMP-9 mRNA levels of all groups dropped to 0.012 ± 0.001 with no significant difference between the ipsilateral cortex and hippocampus and contralateral cortex and hippocampus. In the sham control group, mRNA levels of MMP-9 remained at a relatively constant low value in the ipsilateral cortex (0.011 ± 0.001) and hippocampus (0.010 ± 0.002), and the contralateral cortex (0.009 ± 0.001) and hippocampus (0.010 ± 0.001; p > 0.05).
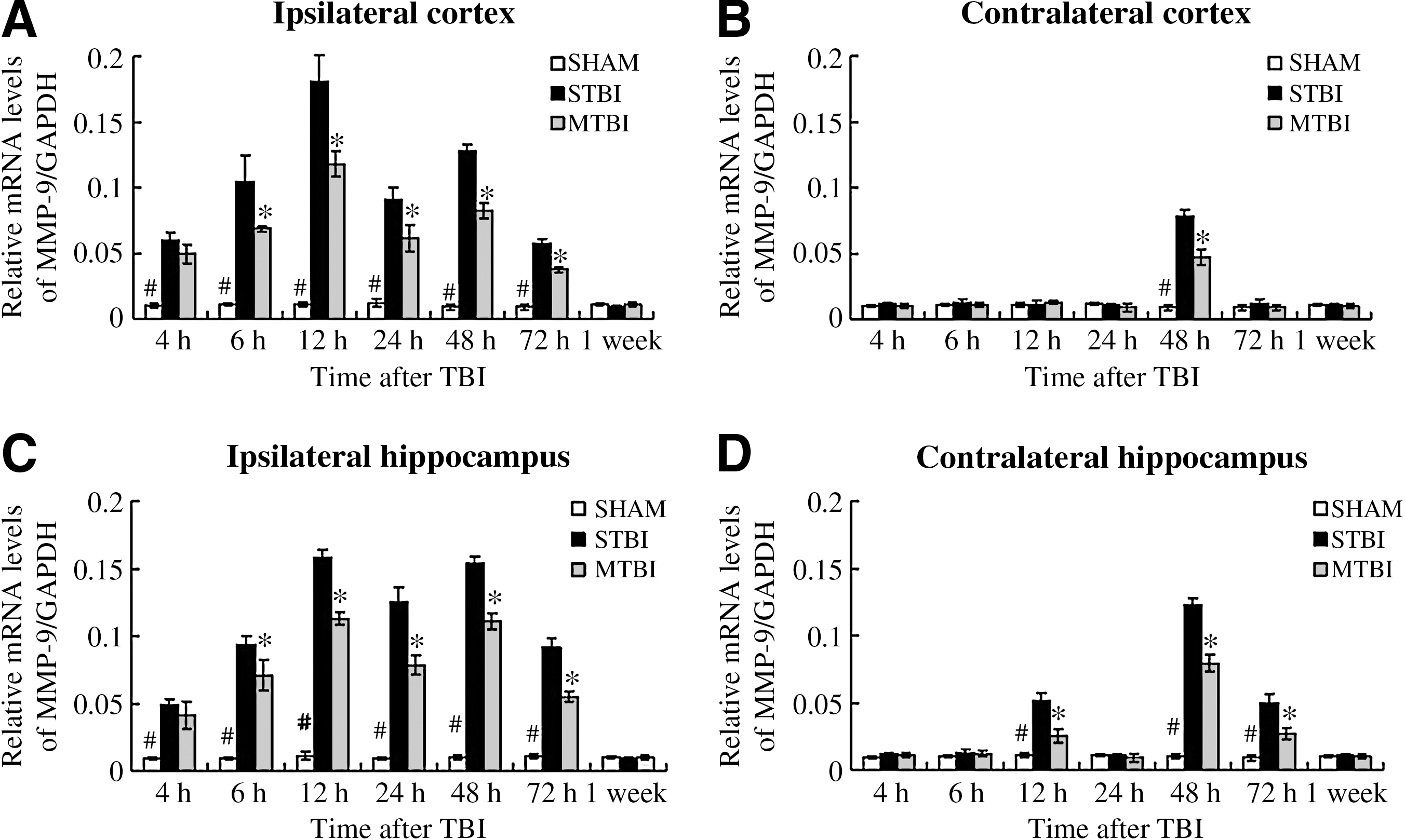
Time course of TBI-induced expression of MMP-9 mRNA. RT-PCR was used to measure relative levels of MMP-9 mRNA in the ipsilateral (
Thus it can be seen that TBI leads to an enhancement in the expression of MMP-9 mRNA compared to the sham group without TBI, and that injury severity determines the degree of elevation of MMP-9 mRNA levels after TBI, both in the injured and uninjured hemispheres.
Temporal and regional expression profiles of MMP-9 proteins after moderate traumatic brain injury
Western blot analysis was carried out to further confirm the expression of MMP-9 protein (Fig. 2). MMP-9 expression was seen in sham-treated animals, indicating that it is present in normal tissues. A relatively constant low level was maintained in the ipsilateral cortex (0.010 ± 0.001) and hippocampus (0.011 ± 0.002), and the contralateral cortex (0.009 ± 0.001) and hippocampus (0.010 ± 0.002; p > 0.05). At 6 h post-TBI, the amount of MMP-9 in both the severe TBI and moderate TBI groups were significantly increased over that of the sham group (p < 0.01). With increasing time, MMP-9 protein levels in the severe TBI and moderate TBI groups kept increasing, with maximal levels reached at 24 h and 72 h after TBI in the ipsilateral and contralateral hemispheres, respectively (Fig. 2). Similarly to the RT-PCR results, a second temporal peak in the amount of MMP-9 protein was seen at 72 h post-TBI in the ipsilateral side. However, for the cortex and hippocampus of the injured (ipsilateral) hemisphere, the increase in MMP-9 protein was only seen in the moderate TBI group, and it was less than that seen in the severe TBI group at all time points (p < 0.01) except 4 h and 1 week. The maximal protein levels of MMP-9 in the moderate TBI group were only 69.28 ± 2.30%, 63.93 ± 1.80%, 60.75 ± 2.80%, and 58.51 ± 2.10% of the corresponding values seen in the severe TBI group in the injured (ipsilateral) and uninjured (contralateral) hemispheres of the cortex and hippocampus, respectively. However, compared with the sham group, the level of MMP-9 protein expression was still significantly higher in the moderate TBI group at all time points except 4 h and 1 week in the injured (ipsilateral) hemisphere (p < 0.01). At 1 week after TBI, MMP-9 protein levels in the injured hemispheres dropped to 0.012 ± 0.001, with no significant differences seen with the contralateral cortex and hippocampus.

Time course of TBI-induced expression of MMP-9 proteins. Shown are Western blots of MMP-9 in the ipsilateral (
MMP-9 mRNA expression at 12 and 48 h after moderate TBI followed by 4 h of post-traumatic temperature manipulation
At 12 h post-TBI, the levels of MMP-9 in the hypothermic groups were significantly decreased compared to those of the normothermic and hyperthermic groups (p < 0.01). However, no significant difference was observed between the values in the hyperthermia and normothermia groups (p > 0.05; Fig. 3).
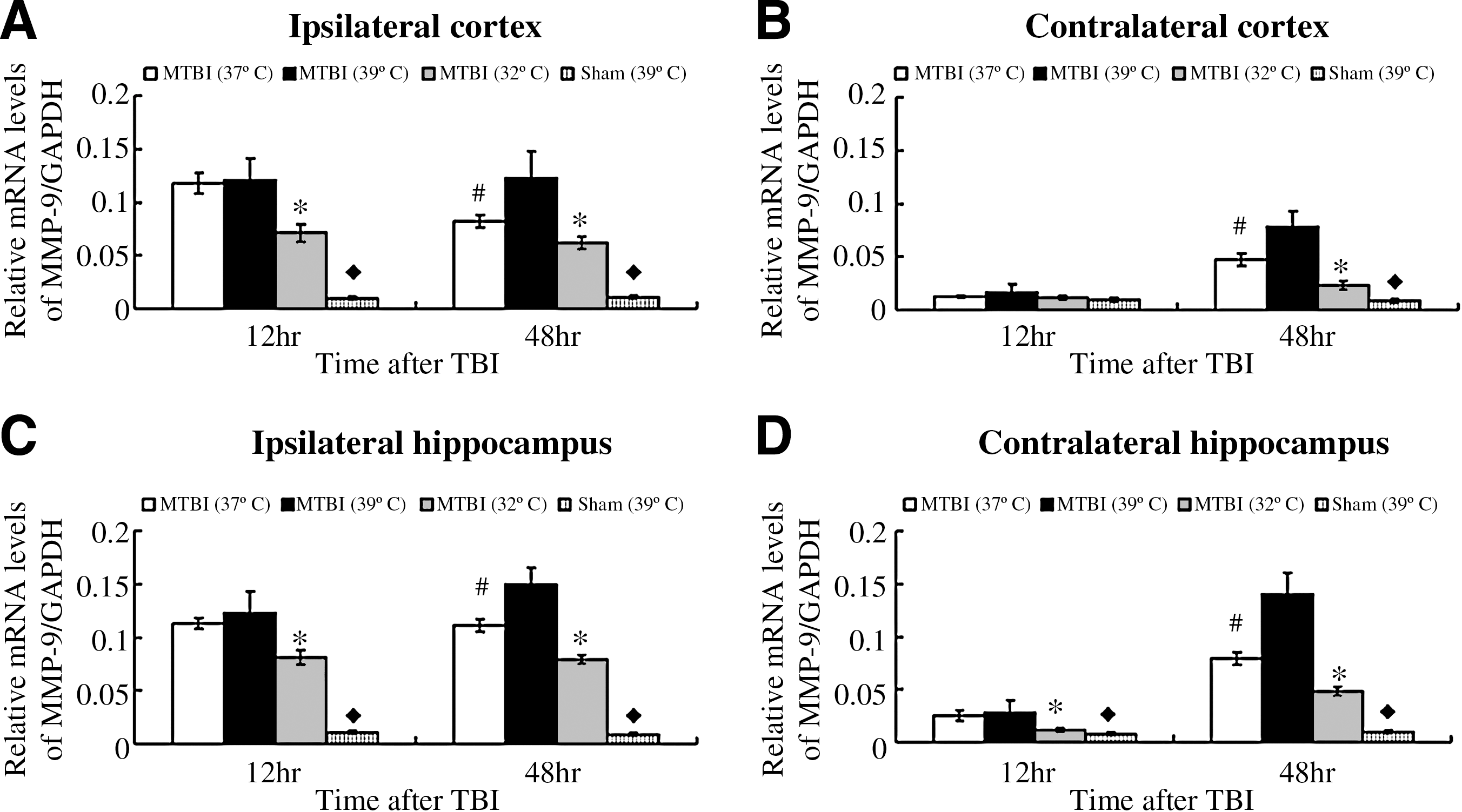
Bar graphs illustrating MMP-9 mRNA expression after moderate TBI, followed by 4 h of post-traumatic temperature manipulation. Normothermia (37°C), hyperthermia (39°C), and hypothermia (32°C), had a significant effect on MMP-9 mRNA expression in the ipsilateral (
At 48 h after TBI, the levels of MMP-9 in the hypothermic groups were decreased significantly over those of the normothermic group, whereas post-traumatic hyperthermia significantly increased (both in ipsilateral and contralateral hemispheres) MMP-9 expression compared with normothermia.
The mRNA levels of MMP-9 remained at a relatively constant low value (p > 0.05). Temperature manipulation alone did not significantly affect MMP-9 mRNA levels for sham-injured groups at 12 and 48 h.
MMP-9 protein expression at 24 and 72 h after moderate TBI followed by 4 h of post-traumatic temperature manipulation
At 24 h after TBI, the MMP-9 protein levels in the hypothermic groups were decreased significantly over those of the normothermic and hyperthermic groups (p < 0.01). No significant difference was observed between the protein levels of the hyperthermic and normothermic groups (p > 0.05; Fig. 4).

Bar graphs illustrating MMP-9 protein expression after moderate TBI, followed by 4 h of post-traumatic temperature manipulation. As shown here, temperature manipulation involving normothermic (37°C) treatment, hyperthermic (39°C) treatment, and hypothermic (32°C) treatment, had a significant effect on MMP-9 protein expression in the ipsilateral (
At 72 h after TBI, the MMP-9 protein levels in the hypothermic groups were decreased significantly over those of the normothermic group. Post-traumatic hyperthermia significantly increased (both in the ipsilateral and contralateral hemispheres) MMP-9 expression compared with normothermia.
The protein levels of MMP-9 remained at a relatively constant low value (p > 0.05). Temperature manipulation alone did not significantly affect MMP-9 protein levels for the sham-injured groups at 24 and 72 h.
Immunofluorescence
The immunofluorescence analyses were performed in the hippocampus of TBI and sham control rats using anti-MMP-9. As can be seen in Figure 5, MMP-9 was co-localized with GFAP in the hippocampus. This result was similar to that observed by Gu and colleagues (Gu et al., 2002).
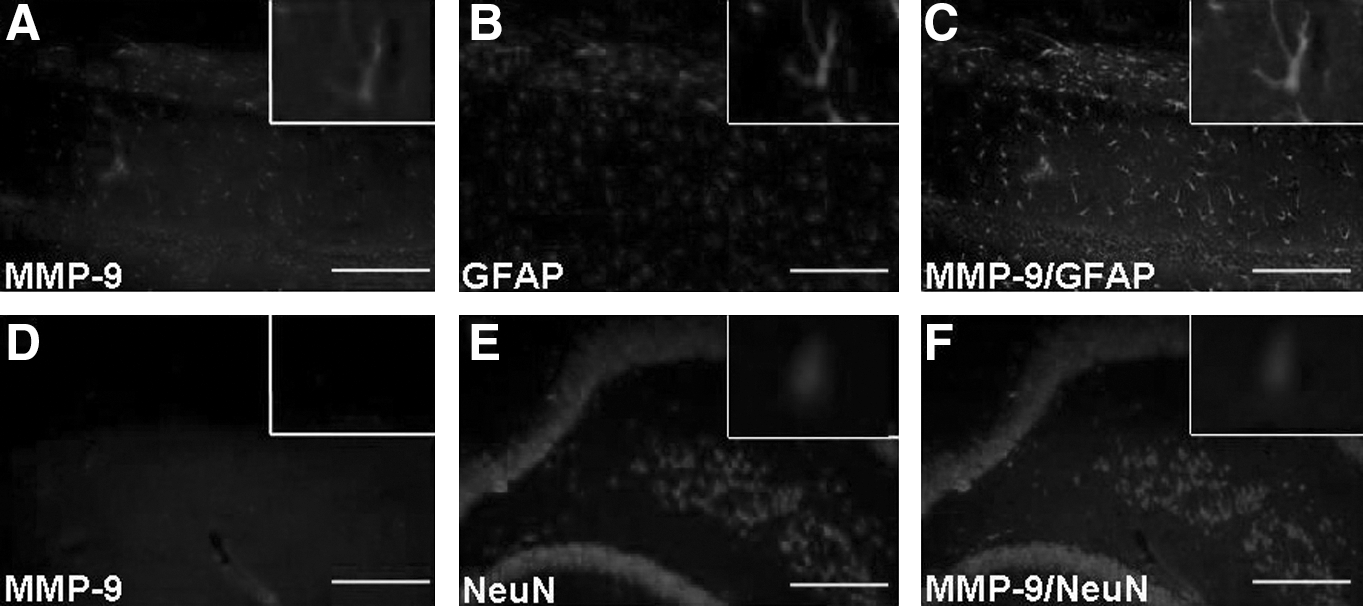
(
Discussion
It was shown in our study that moderate TBI induced significant increases in both MMP-9 mRNA and protein levels in the ipsilateral hemisphere (cortex and hippocampus) within 1 week (p < 0.01). The MMP-9 mRNA and protein expression levels in the ipsilateral hemisphere (cortex and hippocampus) of the severe TBI group were also increased compared with those in the sham group after TBI (p < 0.01), and these increases were significantly enhanced compared with the moderate TBI group. Finally, post-traumatic hypothermia led to a significant decrease in the MMP-9 mRNA levels as measured at 12 and 48 h in the ipsilateral hemisphere (cortex and hippocampus), compared with normothermic levels. The MMP-9 protein levels at 24 and 72 h in these same brain sections were also significantly reduced compared to those of the normothermic group. This is consistent with the finding of MMP-9 levels in arterial blood and internal jugular venous blood in seven patients with TBI (Suehiro et al., 2004). However, post-traumatic hyperthermia resulted in a significant increase in MMP-9 mRNA levels and protein levels (at 48 and 72 h, respectively) in the ipsilateral hemisphere, compared with normothermic levels. This enhancement of the elevation of MMP-9 expression in the post-traumatic hyperthermic group was due to the combined effect of TBI and temperature. These findings suggest that injury severity and temperature are important factors in MMP-9 production after TBI. The protection provided by hypothermia may partially be due to its effect on the downregulation of MMP-9.
It was also shown in our study that MMP-9 protein and mRNA expression profiles were different, not only in different hemispheres, but also in different regions of the same hemisphere with the same level of injury severity. For the moderate and severe TBI groups, two temporal peaks of MMP-9 mRNA and protein expression were observed in the ipsilateral hemisphere (cortex and hippocampus) and contralateral hippocampus, though the first peak value was higher than the second one. However, only one peak in MMP-9 mRNA and protein was observed in the contralateral cortex. This phenomenon suggests that the higher value of the first peak may be due to direct damage, while the lower level of the second peak may be due to the secondary damage caused by autodestructive insults, such as ischemia. The higher MMP-9 values both in mRNA and protein in the contralateral hippocampus (the second peak) than those seen in the contralateral cortex (observed at 48 h and 72 h for mRNA and protein, respectively) may be due to secondary damage (p < 0.05 for mRNA at 48 h, and for protein at 72 h), as the hippocampus is more sensitive to secondary damage such as that caused by ischemia (Chen et al., 2007; Yamashita et al., 2007). The fact that the first MMP-9 mRNA and protein peak values in the ipsilateral cortex were higher than those of the ipsilateral hippocampus may be because the increase seen in the early period (mRNA and protein at 12 h and 24 h, respectively) in the ipsilateral cortex may be due to direct mechanical damage, but the distance between the ipsilateral hippocampus and the damaged site (the ipsilateral cortex) attenuated the increase. This result was similar to that observed by Buss and associates (Buss et al., 2007).
The first description of moderate hypothermia after experimental TBI as a neuroprotective measure in a rat model was reported by Dietrich and colleagues (Dietrich et al., 1994b), while Bramlett and associates and others (Dietrich et al., 1994a) first demonstrated cognitive improvements after TBI with the use of hypothermia (Bramlett et al., 1995). Since then, scientific evidence regarding the effective use of hypothermia has been accumulating, whereas controversy about its role in human patients continues (Peterson et al., 2008). With respect to the physiological effects of body temperature on neutrophil function, it has generally been accepted that mild hyperthermia activates neutrophil function (Johansen et al., 1983; Utoh and Harasaki, 1992; Whalen et al., 1997). Mild hypothermia reduces the synthesis and release of proinflammatory cytokines. Moreover, it was reported that increases in the expression of MMP-9 and MMP-2 were associated with the early inflammatory consequences of injury, including neutrophil and macrophage accumulation after spinal cord injury (Noble et al., 2002). This is also consistent with the results shown in the current work, that hypothermia reduces the synthesis of MMP-9.
It was demonstrated in this study that post-traumatic hypothermia attenuates the increases in MMP-9 mRNA and protein levels, not only for the first peak, but also in the second peak after moderate fluid percussion brain injury. Although previous reports discussed the effects of mild or moderate hypothermia on MMPs and tissue inhibitors of metalloproteinases (TIMPs), such as MMP-9, MMP-2, and TIMP-3 (Jia et al., 2009a; Truentter et al., 2005), less is known about the effects of temperature manipulation on MMP levels after TBI. Jia and colleagues (Jia et al., 2009b) reported that hypothermia after TBI significantly decreased the elevation in TIMP-3 mRNA levels compared with normothermic animals. In contrast to hypothermia, post-traumatic hyperthermia aggravates the histopathological outcomes of TBI (Chatzipanteli et al., 2000; Dietrich et al., 1994a; Jia et al., 2009b). In our study, post-traumatic hyperthermia significantly increased MMP-9 mRNA and protein levels at the second peak (48 and 72 h, respectively) in the ipsilateral and contralateral hemispheres compared with normothermic levels. This suggests that the secondary brain injury resulting from TBI is sensitive to temperature.
MMPs are essential for remodeling the extracellular matrix, tissue morphogenesis, and wound healing (Werb, 1997). However, excessive proteolytic activity of MMPs can be detrimental, leading to numerous pathological conditions, including disruption of the blood−brain barrier (Yong et al., 2001), apoptosis (Gu et al., 2002), and inflammation (Mun-Bryce and Rosenberg, 1998). Overexpression of MMPs was also reported to be important in spinal cord and brain trauma (Noble et al., 2002; Wang et al., 2000). After traumatic spinal cord injury, transient upregulation of MMP-9 due to immunocytochemical changes has been detected (Nobel et al., 2002). It was reported that MMP-9 activity was increased at 3 h post-injury, and persisted at high levels until 7 days after controlled cortical impact injury in rats (Wang et al., 2002). TIMPs are natural MMP inhibitors that impede the proteolytic activity of MMPs by forming noncovalent 1:1 stoichiometric complexes (Fassina et al., 2000). They also regulate MMP and adamalysin metalloproteinase activity. Transient upregulation of TIMP-3 has been detected after traumatic spinal cord injury (Buss et al., 2007). Wallace and associates reported markedly increased TIMP-3 expression in neurons in the cortex and caudate of an ischemic hemisphere (Wallace et al., 2002). In agreement with these findings, TIMP-3 (Jia et al., 2009a) and MMP-9 (Truettner et al., 2005) were upregulated after TBI, and these increases were inhibited by hypothermia. These observations implicate a physiological role for the balance of TIMP-3 and MMP activity at the neuronal level in regulating sensitivity of death receptors.
In this study we report that the production of MMP-9 is dependent on injury severity and post-traumatic brain temperature. As the primary damage resulting from TBI cannot be addressed therapeutically, it is important to optimize strategies to reduce secondary brain injury. Our experiments demonstrated that TBI induces significant increases in MMP-9 mRNA and protein levels in brain tissue. Increases in injury severity led to increases in MMP-9 levels not only in the ipsilateral hemisphere, but also in contralateral brain structures. Subsequent post-traumatic hypothermia significantly attenuated the increase in MMP-9, although more thorough investigations should be carried out on the underlying mechanisms. Thus modest hypothermia, especially in the early phase post-TBI, may be an effective therapeutic strategy to reduce secondary brain injury by influencing MMP-9 expression.
Footnotes
Author Disclosure Statement
No competing financial interests exist.