
Abstract
Diffuse axonal injury (DAI) is a frequent form of traumatic brain injury, and is usually associated with long-lasting neurological impairments. A new experimental model was developed in the present study to induce DAI in rats by combining low linear and angular accelerations. In most clinical scenarios, DAI is caused by these two forms of acceleration in combination. In the injury-producing facility described here, the rat rotated instantly after it had sustained the impact that produced linear acceleration. Rats rotated rapidly 90° in the coronal plane at a peak angular acceleration of 137 ± 12 krad/sec2 with a duration of 33.7 ± 1.2 msec. The linear acceleration was applied to the rat's head by dropping a 450 g weight from a height of 0.9 m. Rats exposed to the combined accelerations took significantly longer to regain consciousness (11.9 ± 3.6 min) than control rats (p < 0.01) or rats subjected to purely angular or linear acceleration (p < 0.01). Although macroscopic damage was observed in all brain-injured animals, axonal damage and hemorrhagic tissue tears were only noted in the animals sustaining the combined accelerations. All rats survived the purely linear or angular acceleration, whereas the mortality rate reached 21.7% following the combined accelerations. These results show that this model is capable of reproducing the major histological and neurological changes that are associated with DAI, and that the combination of low linear and angular accelerations can produce non-linear and synergistic effects to induce moderate/severe DAI.
Introduction
T
The high incidence, morbidity, and cost associated with DAI necessitate a more complete understanding of this condition in order that intervention strategies can be developed and patients can be treated more effectively. Although much effort has been devoted in the past two decades to elucidation of the injury mechanism and the pathophysiological cascades that occur in DAI, no breakthrough in therapy for improvement in outcome has been achieved, and no standard therapy has been supported by Class I evidence (Margulies et al., 2009; Meythaler et al., 2001; M. Smith, 2003; Tolias and Bullock, 2004). In response to this need, the establishment of experimental models of DAI to explore the pathogenesis and to evaluate potential therapies is especially important. These established models are designed to mimic as closely as possible the clinical condition of human DAI, and they have improved our understanding of DAI in certain aspects.
The principal mechanical force associated with the induction of DAI is inertial loading applied to the head (Adams, 1982; Meaney et al., 1995; M. Smith, 2003). He and associates (2000) designed a lateral head rotation acceleration device, in which DAI is produced in rats without a direct impact. In the weight-drop model described by A. Marmarou and associates (1994), DAI is produced in the rat with combined impact and linear acceleration. Although the above studies may replicate DAI through the use of either angular or linear acceleration alone, there is rarely a trauma due to purely rotational or linear acceleration of the head in realistic accidents (Huh et al., 2007; King et al., 2003; Mclean, 1995; Prange et al., 2003; Ueno and Melvin, 1995). In addition, the angular acceleration is generally believed to be the main condition responsible for DAI (Gutierrez et al., 2001; D.H. Smith et al., 2003; J. Zhang et al., 2006), even though it has been realized that most patients with TBI sustain both forms of acceleration in combination (Duma et al., 2005; King et al., 2003; Yoganandan et al., 2009). If angular acceleration is the major factor that contributes to the generation of DAI, then what role does the linear acceleration play, and what will be the effects when linear and angular accelerations are combined? The development of a model that can combine both forms of the acceleration is therefore critical for the study of the underlying mechanisms of DAI and the subsequent development of specific preventative measures.
The purpose of the present study was to develop and characterize a new experimental model in which moderate/severe DAI could be induced consistently in rats through a combination of low linear and angular accelerations. Having established this new experimental model, we considered the biomechanical, neurological, and histological changes that occurred in this rat model of DAI. Neurological and histological alternations were also compared with those produced by purely linear or angular acceleration, and then the combinatorial effects were discussed.
Methods
All protocols that involved the use of animals were approved by the Animal Use Committee of Shanghai Jiao Tong University School of Medicine, and were in compliance with the Guide for the Care and Use of Laboratory Animals.
Design of the experimental model
The experimental model consisted of a linear acceleration system and a coronal plane angular acceleration system (Fig. 1). The impact-induced linear acceleration system was an adaptation of that used in Marmarou's model (A. Marmarou et al., 1994). A ring stand with clamps supported a Plexiglas tube and a pulley; the pulley was fastened above the Plexiglas tube. A 450-g cylindrical weight that was attached to one end of a non-elastic rope fell freely from a designated height, guided by the Plexiglas tube onto a metallic helmet fixed to the skull vertex of the rat by dental acrylic. The other end of the non-elastic rope, which was directed over the pulley, was connected to a trigger button in the angular acceleration system. In addition, a 5.5-cm-thick foam bed that supported the rats was pasted firmly onto a plate that extended laterally from the angular acceleration system. The angular acceleration system was adapted from the lateral head rotation model designed by He and associates (2000). This rotational system consisted of the following parts: A support mechanism that included a laterally extended plate and three belts for binding. A driving spring (16 coils, 5.0 cm inside diameter) made of a high-quality 4-mm-thick steel wire, which was pretwisted 270° and then affixed to the rotational bearing. Interlocks that consisted of a spacer pin, trigger button, non-elastic rope, and trigger spring. The combination of linear and angular accelerations was carried out through the interlocks. Rotation angle-determined block, which assured reliable selection of the desired rotational angle (rotation was limited to 90° in the present study).

Diagram of the experimental device for producing DAI by the combination of linear acceleration and lateral head rotation. 1 = pulley; 2 = cylindrical weight; 3 = non-elastic connective rope; 4 = Plexiglas tube; 5 = laterally extended plate; 6 = spacer pin; 7 = trigger spring; 8 = trigger button; 9 = driving spring; 10 = rotation angle-determined block; 11 = rotational bearing; 12 = foam bed.
The injury-producing device was affixed atop a heavy, stable laboratory table.
Induction of diffuse axonal injury
Adult male Sprague-Dawley rats (n = 108) weighing 300–350 g were used for this study. They were maintained with dark/light cycles of 12 h duration, and food and water were available ad libitum. Animals were anesthetized by intraperitoneal injection with 3% sodium pentobarbital (60 mg/kg body weight) and were allowed to breathe spontaneously. A midline incision exposed the dorsal surface of the skull, upon which a steel disc was cemented centrally between lambda and bregma using dental cement. Then, the laterally extended plate with the foam bed was confined to a horizontal position (at which time the drive spring had already been twisted 360°), the rat was placed in a prone position on the foam bed, and the head of the rat was located close to and in line with the rotational bearing. When the position of the rat had been established, two belts were fastened around the trunk of the rat to prevent the animal from falling off the foam bed after the induction of trauma, and a third belt was fastened around the head of the animal to maintain the alignment between the center of rotation of the head and the rotational bearing. The lower end of the Plexiglas tube was positioned directly above the helmet. After the point of contact between the weight and the helmet had been targeted, the impact was delivered by dropping a 450-g weight from a height of 0.9 m when the rat lost its corneal reflex. Due to the fact that the weight continued to drop a further 0.01 m after contacting the helmet, the non-elastic rope pulled the trigger button, the spacer pin was shifted by the trigger spring, and the rat was then rotated rapidly clockwise by 90°. The rotation was limited by the rotation angle-determined block. A “second hit” was avoided because the rat had been displaced from the original position after rotation. Following brain injury, the metal disc was removed and the midline incision wound was closed. The animals were subjected to four types of injury: group 1 animals were subjected to the combined linear and angular accelerations (n = 60); for group 2 animals, a 450-g weight was dropped from a height of 0.9 m with no subsequent rotation (n = 18); group 3 animals underwent purely angular acceleration with no preceding direct impact (n = 18); group 4 animals served as the control (n = 12). The latter sham-operated control animals underwent the above procedures except for the impact and rotation. No resuscitation was performed, and the mortality was assessed at 24 h post injury. Each group was divided randomly into six subgroups, which were assessed histologically at different time points.
Measurement of rotational kinematics
Biomechanical parameters were measured and calculated under the supervision of the National Die & Mould Engineering Research Center, Shanghai Jiao Tong University School. Test instruments included a rotary encoder (S40-6-3600ZO; Mecapion, Xinqu, China), a programmable logic controller (PLC; FX2N-16MR-001; Mitsubishi Electric, Tokyo, Japan), a text display (MD204L; Step Servo, Shenzhen, China), and a high-speed camera (HiSpec; Peiport Scientific, Quarry Bay, Hong Kong). The rotary encoder was affixed to the rotational bearing, with which the center of rotation of the rat was aligned. The rotary encoder, with a rotation capability of 3600 pulses per cycle, emitted a pulse each time the angular acceleration system rotated 0.1°. Therefore, when the angular acceleration system had completed 90° rotation, the PLC received 900 pulses. Through the photocoupler and timer of the PLC, the PLC was programmed to start timing when it had received one pulse and to stop timing when it had received a total of 900 pulses. The rotation time was calculated from this interval. Angular velocity and angular acceleration were numerically integrated from the number of pulses detected in the corresponding time. The number of pulses was tracked and detected per millisecond using the open-collector output circuits between the rotary encoder and the PLC. In accordance with a series of mathematical formulas, the kinematic equations were shown as follows:
where ω is the angular velocity and P is the number of pulses per millisecond.
where α is the angular acceleration and N is the difference in number of pulses between two successive milliseconds. Finally, the calculated values were shown on the text display, and the peak value was selected. Given that the grating plate of the rotary encoder and the head of the rat rotated around the same axis in the process of rotation and that their rotational radii were similar, theoretically, the rotation time, angular velocity, and angular acceleration during rotation of the rat's head and the rotary encoder should be the same. The tests of rotational kinematics were conducted in 24 rats using an identical experimental setup. A high-speed camera was used to determine the interval between the impact of the weight and the subsequent rotation, and to validate further the reliability and reproducibility of the device. The duration of the pure impact in group 2 was also measured with the high-speed camera.
Neurological and histological observations
The rats in the experimental group sustained injury when they lost their corneal reflex. The latency to recovery of other reflexes was also assessed from this time point. The reflexes were assessed according to the methodology of Fijalkowski and associates (2007). The duration of suppression of the corneal reflex was used as an index of traumatic unconsciouness. The brains of rats that died prematurely post injury were removed immediatedly after death and examined for gross macroscopic changes. At assigned time points (1, 6, 12, 24, 72 h, and 1 week post trauma), the animals were anesthetized deeply with an overdose of 3% sodium pentobarbital (90 mg/kg body weight administered intraperitoneally), and prepared for histological and morphological analysis. Briefly, the thoracic cavity was opened, a perfusion catheter was inserted into the ascending aorta, the right atrium was incised, and the rat was perfused transcardially with 100 mL of isotonic saline to clear blood from the vasculature. This was followed by perfusion with 350 mL of 4% paraformaldehyde at 4°C for immunohistochemistry. Two rats from each subgroup of group 1 were perfused alternatively with 350 mL of 2.5% glutaraldehyde at 4°C for electron microscopy. The entire brain was removed for gross observation, cut into 3-mm-thick coronal blocks, and immersed immediately in the same fixative at 4°C for 24 h. It was then dehydrated routinely and embedded in paraffin. Serial coronal sections were then cut (10-μm-thick sections for hematoxylin and eosin [H&E] staining and immunohistochemistry). The brainstem was bisected in the median sagittal plane, then blocked sagittally, and sectioned. The sections were incubated overnight at 4°C with a rabbit antibody against the C-terminus of beta-amyloid precursor protein (β-APP; 1:300; AbCam, Cambridge, UK) to determine the presence of axonal injury. Binding of the primary antibody was detected by incubating the sections with a biotinylated swine anti-rabbit secondary antibody (1:400; Dako, Glostrup, Denmark) for 30 min at 37°C, and visualizing with streptavidin-peroxidase (1:400; Dako) and diaminobenzidine (DAB kit; Boster, Wuhan, China) as the chromogen. The sections were then counterstained with haematoxylin, dehydrated in graded alcohols, cleared in xylene, and mounted. To control for non-specific binding of the secondary antibody to endogenous sites within the tissue, immunohistochemical controls were performed in which the primary antibody was omitted. Blocks of 1-mm3 tissue prepared for transmission electron microscopic (TEM) was osmicated, dehydrated through graded ethanol, and embedded in Epon 618 (Tianyuan Group Shanghai Resin Factory Co., Ltd., Shanghai, China). Semithin sections were cut from the blocks. The changes in axonal ultrastructure were evaluated using a Philips CM-120 TEM (Philips, Eindhoven, Netherlands).
Histological analysis
Although termed “diffuse,” the pattern of axonal damage after DAI is more accurately described as multifocal since it occurs in discrete areas throughout the deep and subcortical cortical white matter, and is particularly common in midline structures, including the corpus callosum and brainstem (Adams, 1982; Adelson et al., 2001; Povlishock and Christman, 1995). Therefore, these latter two regions were chosen for the quantification of damaged axons. Sections of the corpus callosum were located 3.8 mm posterior to bregma. Sections of the brainstem were obtained from a site 1 mm off the sagittal midline. Two slides were chosen for each animal, one from each of the above two sets of sections, and immunoreactive axonal swellings/bulbs in the selected regions were counted in four consecutive microscopic fields at a magnification of 200 ×. The results of previous experimental studies have suggested that the hippocampal CA3 region is selectively vulnerable to TBI (Baldwin et al., 1997; Clark et al., 1997; Grady et al., 2003). Cell counts were performed for the pyramidal layer of CA3 to assess neuronal loss at 1 week after brain injury. Two H&E-stained slides per animal (bregma −3.8 mm) were chosen randomly, and cells were counted in four consecutive microscopic fields at 400 ×. Neurons were counted only if there was a clearly defined cell body and nucleus. All counting was processed by one experienced neuropathologist who was blinded to the injury status of each animal.
Statistical analysis
All continuous data are expressed as the mean ± standard deviation (SD). Average latencies to the recovery of consciousness among the four groups, the peak number of immunoreactive axons among the different regions, and histological cell counts among the four groups were compared using one-way analysis of variance (ANOVA) followed by Newman-Keuls post-hoc tests. Differences were considered significant at p < 0.05.
Results
Biomechanical parameters
Data on the rotation time, angular velocity, and angular acceleration of the device in the presence and absence of a rat are shown in Table 1. After the rat had sustained the impact and linear acceleration caused by dropping a 450-g weight from a height of 0.9 m, the animal experienced two phases in the process of 90° rotation. The first phase was a sudden acceleration caused by the driving spring, during which velocity increased gradually and maximum acceleration was at the initial rotation (their peak values were 70.2 ± 3.7 rad/sec and 15.9 ± 1.7 krad/sec2 respectively). In this phase, animals also sustained linear acceleration during rotation because of inertia after the weight-drop impact. The second phase corresponded to an instant deceleration and stopping when the fixation mechanism crashed into the rotation angle-determined block (the deceleration was up to 137 ± 12 krad/sec2). The coefficient of variation of the peak angular acceleration was 8.75%, demonstrating a high level of consistency of the biomechanical parameters. These peak values were much smaller than those obtained in He's model (He et al., 2000). The angular rotation was triggered as soon as the weight had dropped a further 0.01 m after making contact with the helmet of the rat through the interlocks. The time between the weight making contact with the helmet and the subsequent angular rotation was 17.3 ± 0.5 msec. This small coefficient of variation of 2.89% demonstrated the repeatability of the model described herein. The duration of the pure impact in group 2 was 16.9 ± 0.7 msec, which was close to the above-measured time interval (17.3 ± 0.5 msec) in group 1. This consistency further showed the reliability of the present model. There were no statistical differences with respect to rotational parameters between groups 1 and group 3 (data not shown). Variation in rat weight had minimal influence on the forced response, as checked by recording the acceleration with varied rat weight in the experimental group (data not shown).
Values are presented as mean ± standard deviation.
Acute neurological evaluation and mortality
After purely linear or angular acceleration, all the rats survived, whereas the combination of linear and angular accelerations resulted in a mortality rate of 21.7%. Although most of the experimental rats survived, they all experienced apnea that lasted for several seconds and a reduction in respiratory rate. Seizures only occurred in those rats subjected to the combined accelerations, and were limited to the immediate post-injury period. Subsequently, respiration in the brain-injured rats recovered gradually. The rats that did not survive experienced extended apnea, which lasted for up to 20 sec, and their respiratory rate reduced gradually until death; these deaths occurred from 3 to 92 min post injury. All brain-injured rats manifested post-traumatic physiological and behavior changes, which were characterized by several seconds of apnea, deep and slow respiration, prolonged latency of recovery of reflexes, and absent or slow movements. Animals that sustained a single type of acceleration demonstrated normal ambulation after 3 h post injury; however, the animals exposed to the combined accelerations took 6 h to recover normal ambulation. The latency of recovery of the corneal reflex was significantly longer in rats under the combined accelerations (11.9 ± 3.6 min, p < 0.01; Fig. 2) than in control rats (6.5 ± 1.3 min) or in rats under the angular (8.5 ± 2.3 min) or linear acceleration (6.9 ± 1.8 min). In addition, the latency to the recovery of consciousness also showed a significant difference between the angular acceleration and control rats (p < 0.05). No significant difference was found among other groups (p > 0.05). The recovery times for the other reflexes are presented in Table 2.

Bar graph showing effects of different types of injury on the latency of recovery of the corneal reflex. Animals following combined accelerations showed significantly (# p < 0.01) longer latency compared with all other groups. The latency also showed a significant difference between angular acceleration and control rats (*p < 0.05). No significant differences (p > 0.05) were found among other groups. Data are presented as mean ± standard deviation, and error bars represent standard deviation.
Data are presented as mean ± standard deviation.
Pathological changes: Histology, immunohistochemistry, and electron microscopy
Skull fracture was absent in all rats. Gross morphological observation of the injured brains showed an absence of contusions or focal lesions. Although subarachnoid hemorrhages were observed in all brain-injured rats, blood was present to a limited extent over the brain surface following a single type of acceleration, and hemorrhagic tissue tears were observed separately in the brain parenchyma following the combined accelerations (Fig. 3). Petechial hemorrhages that were dispersed widely in the brainstem, internal capsule, corpus callosum, and subcortical white matter were also noted frequently in the animals subjected to the combined accelerations. Animals that died showed severe epidural hemorrhage and blood pooling at the basal cisterns and cisterna magna. These observations indicated that direct tearing of tissue occurred when the brain was subjected to the combined accelerations. Histological examination of H&E-stained sections revealed the presence of shrunken neurons associated with perineuronal vacuolation and tissue rarefaction in all brain-injured animals, in particular those located dorsal to the corpus callosum and cingulum, as well as the hippocampus (Fig. 4). However, the degree was more pronounced in the animals suffering the combined accelerations. Axonal swellings associated with intraparenchymal hemorrhages were also only observed in these animals; at the locus of discontinuity of axons, the proximal end formed a bulblike protrusion (Fig. 4B). These traumatic changes were significant at 24 h post injury. No similar changes were revealed in the brains of control rats (Fig. 4F). In all brain-injured animals at 1 week post injury, there was a marked drop-out of neurons in the hippocampal CA3 region, as well as overt signs of disrupted cellular alignment (Fig. 5). Reduced neuronal density and cell loss were most extensive and conspicuous following the combined accelerations.
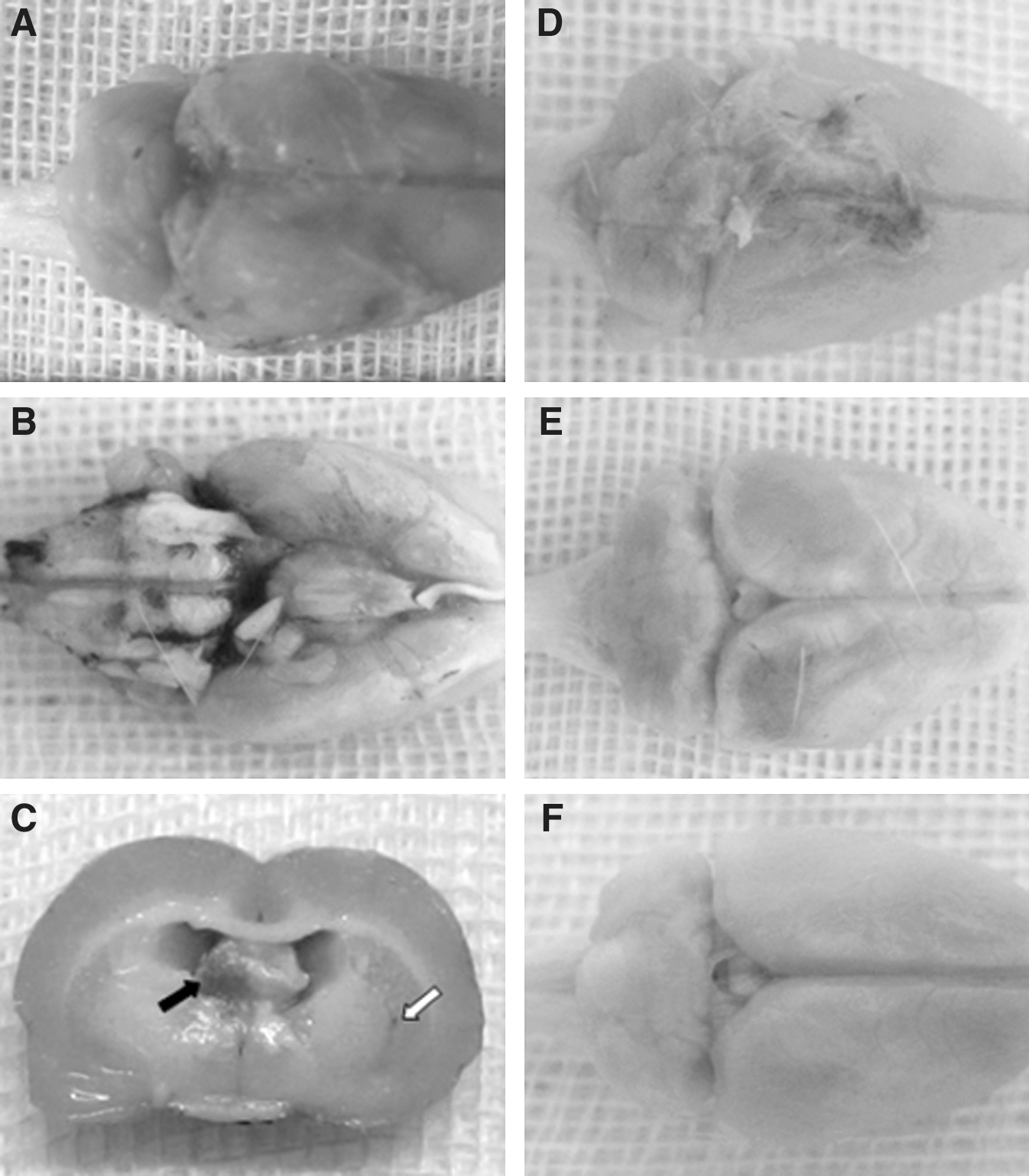
The injured brains of rats at 24 h after combined accelerations: superior surface (

Paraffin sections stained with hematoxylin and eosin (H&E). Representative photomicrographs of the cortex (
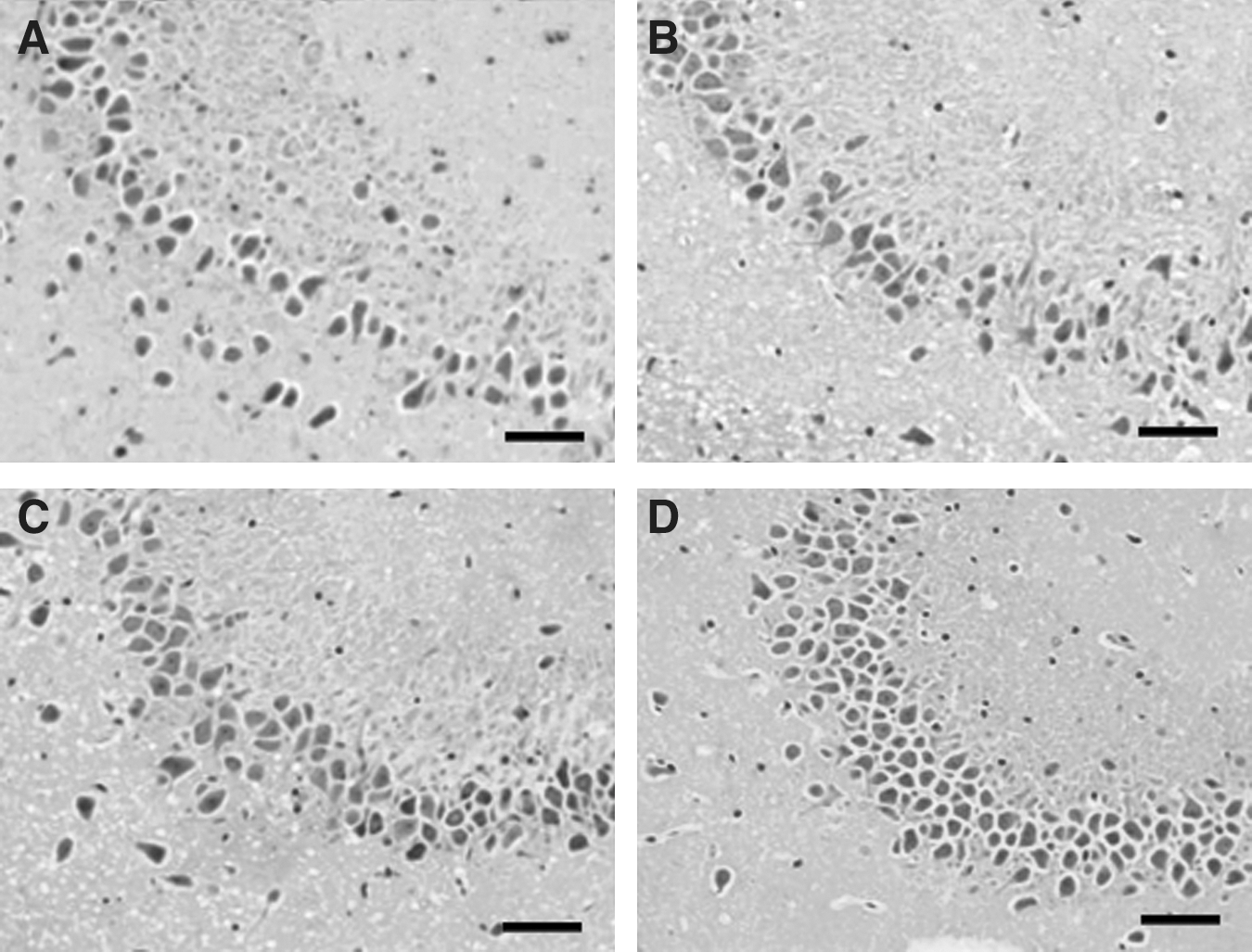
Photomicrographs showing the results of hematoxylin and eosin (H&E) stained sections in the hippocampal CA3 region. At 1 week after trauma, a drop-out of neurons as well as disrupted cellular alignment was apparent in all brain-injured animals following combined accelerations (
Immunohistochemistry to detect β-APP revealed the presence of spheroidal axonal swellings and axonal twisting in many brain regions following the combined accelerations, particularly in the brainstem (Fig. 6A–C). In addition, there was also a profound increase in β-APP immunoreactivity in neuronal cell bodies (Fig. 6D). The increase of β-APP in neurons has also been noted in other models of diffuse TBI, and is thought to represent an acute-phase traumatic-stress response post injury (Cernak et al., 2004; Kilbourne et al., 2009; Van Den et al., 1999). At 6 h post injury, immunoreactivity for the β-APP demonstrated the punctuate and tubulovesicular morphology of axons; however, the majority of these axonal profiles had progressed to form the classically described bulbous axonal swellings, typically referred to as axonal retraction bulbs (Gennarelli et al., 1982; C.R. Marmarou et al., 2005; Maxwell and Graham, 1997). By 72 h post injury, β-APP immunostaining had localized numerous fine deposits (Fig. 6C). No additional axonal damage was demonstrated in sham-injured brains or brains that received a single type of acceleration by β-APP immunohistochemistry (Fig. 6F–I). As illustrated in Fig. 6F, some neurons in the hippocampus were stained slightly following the purely angular acceleration; however, no stained neurons were identified in other regions (Fig. 6G), which is indicative of the vulnerability of hippocampal neurons following TBI (Geddes et al., 2003).

Photomicrographs demonstrating the results of immunohistochemical testing for β-amyloid precursor protein (β-APP). Traumatically injured axons (solid arrow) and neurons within the brainstem showing immunoreactivity following combined accelerations (
Examination of H&E-stained sections revealed a significant loss of cellular profiles in the hippocampal CA3 region at 1 week in the animals subjected to either the purely angular acceleration or the combined accelerations compared with that seen in the control animals (Fig. 7; p < 0.01). Significantly more neurons were lost following the combined accelerations than the purely angular or linear acceleration (p < 0.01). There was no significant difference between the two single-acceleration groups for this measure (p > 0.05). Quantitative analysis, as assessed by β-APP immunohistochemistry, revealed that the mean number of immunoreactive axons in the midbrain increased from 11.7 ± 2.3 at 1 h to 27.3 ± 4.7 at 6 h, and peaked at 12 h (38.6 ± 7.5). The mean number of immunoreactive axons in the medulla oblongata displayed 5.6 ± 1.8 at 1 h that increased to 12.3 ± 2.4 at 6 h and peaked at 24 h (22.5 ± 5.3). The progression of immunostaining axons in the corpus callosum demonstrated a similar trend to that in the medulla oblongata (Fig. 8). The peak number of damaged axons in the midbrain was significantly more than that in the medulla oblongata or corpus callosum (p < 0.01; Table 3).
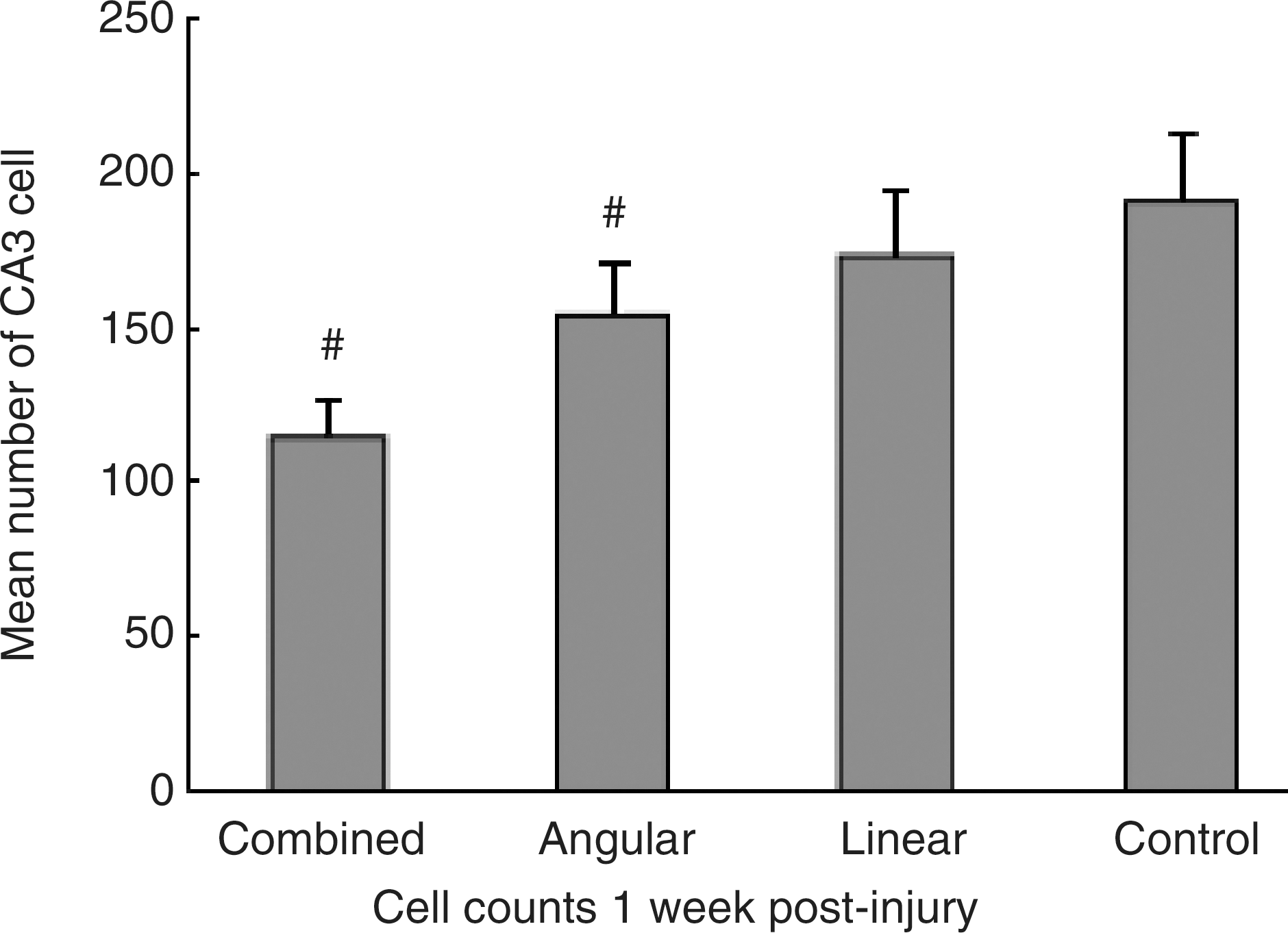
Bar graph demonstrating numbers of cell in the CA3 subfield of the hippocampus at 1 week following combined injuries, single injury, and sham injury. Significant cell loss could be detected in the animals subjected to either combined accelerations or angular acceleration compared to control animals (# p < 0.01). The loss of neurons following combined accelerations was significantly more than that seen in linear or angular acceleration (# p < 0.01). There was no statistical difference between the two single-acceleration groups for this measure (p > 0.05). Data are presented as mean ± standard deviation, and error bars represent standard deviation.

Line chart showing the different trends of progression of the damaged axons in different regions following combined accelerations. The mean number of immunoreactive axons in the midbrain peaked at 12 h post injury. However, immunoreactive axons in the medulla oblongata and corpus callosum both peaked at 24 h post injury. Data are presented as mean ± standard deviation, and error bars represent standard deviation.
The peak number of damaged axons in midbrain is significantly (p < 0.01) more than that in corpus callosum and medulla oblongata. No significant difference (p > 0.05) is revealed between corpus callosum and medulla oblongata. Data are presented as mean ± standard deviation.
The above sequences of morphological changes in the injured axons following combined accelerations (axonal constriction and swelling, axonal disconnection, and formation of axonal retraction bulbs) were visualized more readily at the EM level. Moreover, electron microscope (EM) demonstrated the presence of dramatic protuberances of the axolemma, multiple segmentations of the axonal cylinder, and accumulation of organelles in the distended axons, as well as the loss of microtubules and neurofilament compaction (Fig. 9). Two different types of axonal injury were revealed. The first type exhibited vacuolization and concomitant disruption of the surrounding myelin sheath (Fig. 9A). However, these dramatic morphological changes were not associated with an overt dissolution of the cytoskeleton. Rather, they appeared in conjunction with linearly aligned and compacted neurofilaments. The second type presented significant axonal swelling and dissolution of the cytoskeleton, in association with the accumulation of organelles (Fig. 9D). The second finding corresponded to the observations for the β-APP-immunolabeled axons (Fig. 6B). These two different classes of traumatic axonal injury described in the current investigation are consistent with previous studies (DiLeonardi et al., 2009; C.R. Marmarou et al., 2005; Stone et al., 2001).

Electron micrographs demonstrating different pathological changes and different types of axonal injury in the corpus callosum at 24 h following combined accelerations. (
Discussion
DAI, clinically associated with loss of consciousness and extended deep coma after TBI, makes a major contribution to disability in approximately 40% of closed head injuries (Buki and Povlishock, 2006; Meythaler et al., 2001). The principal mechanical force associated with the induction of DAI is inertial loading applied to the head, which occurs commonly in road traffic accidents (Adams, 1982; Gennarelli et al., 1998; D.H. Smith et al., 2003). This inertial force induces dynamic shear, tensile, and compressive strains within the brain tissue, which result in dynamic tissue deformation. Based on this principle, several models of DAI have been developed to explore the underlying injury mechanisms. Of these, the weight-drop model of A. Marmarou and associates (1994) and the lateral head rotation model of He and associates (2000) have been used frequently to induce DAI in rodents. Linear acceleration with impact and rotational acceleration are applied respectively in these two models. However, analyses of real-time measurements of head accelerations have revealed that football players and boxers often sustain combined linear and angular accelerations in the process of head-impact events (Duma et al., 2005; Guskiewicz et al., 2007; Rowson et al., 2009; Waliko et al., 2005). Because of small acceleration pulse durations, severe brain injuries seldom occur in football players and boxers (Fijalkowski et al., 2007; King et al., 2003). Although it is difficult to establish whether clinical patients from traffic accidents or falls experience both linear and angular accelerations because of the difficulty in obtaining simultaneous medical and crash-related data, a growing body of evidence suggested that most patients with TBI suffer direct impacts (Huh et al., 2007; King et al., 2003; Mclean, 1995; Prange et al., 2003; Yoganandan et al., 2009). In addition to inducing linear acceleration, a direct impact also usually imparts rotational kinematics to the brain because of factors such as head shape, eccentric line of action of the contact force, and skull-bone characteristics (Vander Vorst et al., 2007; Yoganandan et al., 2009). So, clinical patients with TBI are inclined to sustain a combination of linear and angular accelerations (King et al., 2003).
Axons are supple under normal conditions, and they become brittle when exposed to the rapid deformations associated with brain trauma; the production of rapid shear deformations needs fairly strong inertial forces (Bayly et al., 2006; D.H. Smith and Meaney, 2000). The magnitude of acceleration needed to cause brain injury is inversely related to brain mass, and extremely high levels of acceleration are required to produce axonal damage in small animals (Duhaime, 2006; Margulies and Thibault, 1989). Therefore, it would be difficult to induce severe DAI in rodents purely by increasing the magnitude of the linear or angular acceleration (Fijalkowski et al., 2007). Moreover, Ueno and Melvin (1995) indicated that the use of either translational or rotational motion alone to study head injury may not only result in an underestimation of the severity of an impact but may also prevent the identification of critical sites of injury in the brain. King and associates (2003) further suggested that moderate levels of these two forms of acceleration in combination can often cause severe brain injuries. Unfortunately, in existing models of DAI, including the two models mentioned above, head injury is induced using purely linear or angular acceleration, even though such impact conditions do not usually occur in real life. The results of the present study demonstrate that linear and angular accelerations can be combined in this new clinically relevant experimental model to induce moderate/severe DAI at low values of acceleration. The interlocks, which are key features to the present model, allow the rat to experience instant rotation after it has sustained weight-drop impact.
The animal experimental model presented in this study was based on both Marmarou's weight-drop model and He's lateral head rotation model. Although both linear and angular acceleration can individually cause DAI, as these two models have demonstrated, the level of biomechanical parameters needed to produce moderate/severe DAI is very high. Producing moderate/severe DAI required a 450-g weight to be dropped from a height of 2 m in Marmarou's model, in which there was also the possibility of a “second hit” due to the weight rebounding from the skull. The angular acceleration of rat lateral head rotation in He's model was up to 1.806 × 105 rad/sec2. In the present investigation, the animals subjected to either purely linear acceleration, by dropping a 450-g weight from a height of 0.9 m, or purely angular acceleration of approximately 1.37 × 105 rad/sec2 demonstrated no evidence of axonal damage. However, since combined linear and angular accelerations at the above levels resulted in moderate/severe DAI, a remarkable additional insight gleaned from these studies is that the combination of linear and angular accelerations can produce non-linear and synergistic effects. The combinatorial effects may explain why DAI is a common phenomenon in clinical TBI, and is not readily reproduced purely by acceleration in experimental TBI.
Rotational kinematics are generally considered to be the main factor responsible for DAI (Adams, 1982; Gennarelli et al., 1998; J. Zhang et al., 2006). However, recent studies suggested that high non-impact rotational acceleration alone may not be sufficient to produce traumatic axonal injury (Prange et al., 2003). Moreover, some animal models and finite element models have demonstrated that linear acceleration can also induce DAI (A. Marmarou et al., 1994; Nishimoto and Murakami, 1998). Considering the above results and the combinatorial effects demonstrated in the present study, it may be concluded that the generation of DAI should not be linked to angular acceleration alone, and that the combinatorial effects of linear and angular accelerations may play a pivotal role in the production of DAI in the clinical situation. It has also been revealed in a finite element model that the effect of tangential motion is enhanced significantly when translational and rotational accelerations are combined (Ueno and Melvin, 1995). These conclusions may explain how the model described herein could induce moderate/severe DAI through combining low values of linear and angular accelerations.
In addition to consistent input biomechanics, this study produced the functional and pathological outcome that was consistent with that of DAI in humans. Animals exposed to the combined linear and angular accelerations exhibited a prolonged duration of coma, which was longer than that of animals subjected to a single type of acceleration, and was accompanied by physiological changes and behavioral suppression. A single type of acceleration resulted in no mortality; surprisingly, the post-traumatic mortality rate reached 21.7% following the combined accelerations, suggesting severe TBI. Hemorrhagic tissue tears, which are recognized as being a characteristic feature of a severe form of DAI (Graham et al., 2000), were observed in the animals sustaining the combined accelerations. Multiple petechial hemorrhages in the white matter were also noted in the present model. However, in Marmarou's model, petechial hemorrhages were limited almost exclusively to the brainstem (Foda and Marmarou, 1994). The absence of petechial hemorrhages in other regions, such as the corpus callosum, might be associated with the mechanics of the injury applied in Marmarou's model, where the motion of the animal was only oriented in the sagittal plane and the deflection of the corpus callosum was small. If motions were applied in the coronal plane, the function of the cerebral falx would be significant, and the shear stresses would be concentrated on the corpus callosum (L. Zhang et al., 2001).
Traumatic axonal injury (TAI), the pathological hallmark of DAI, was only demonstrated in the animals exposed to the combined accelerations. Sphaeroid and twisted structures of axons were noted in multiple regions, which mainly involved the brainstem, corpus callosum, subcortical white matter, and hippocampus. These axonal structures corresponded to the accumulation of β-APP at sites of axonal swelling and retraction bulbs. In accordance with previous descriptions of TAI by C.R. Marmarou and associates (2005) and Wilkinson and associates (1999), who reported a correlation of survival time with size of axonal swellings in DAI, we also observed an increase in the size of axonal bulbs over time. Quantitative analysis revealed that the time for the progression of damaged axons of different regions was heterogeneous. This may be a result of differing severities of injury in different regions. The extent of axonal damage in the midbrain and corpus collosum was more serious than that in the medulla oblongata in our study, whereas the spatial characteristic was just the reverse in He's model (He et al., 2000). This may be explained by the fact that the center of rotation of the rat in our model was aligned with the vertebral column. However, the rotation of the rat's head in He's model was induced relative to the neck. Therefore, the biomechanical effects of the shearing force in He's model were focused on the part of the brain located primarily in the atlantooccipital area, and the upper cervical spinal cord and medulla oblongata sustained the most severe injury. In Marmarou's model, the brainstem was accelerated severely down into the skull base with 2 m level injury, and predominant axonal damage was located in the brainstem and cortical areas under the site of impact (Foda and Marmarou, 1994; Kilbourne et al., 2009). It has been demonstrated in a finite element model that a high shear stress concentration was generated at the brainstem when a sagittal section of the head was subjected to an impact (L. Zhang et al., 2004).
There is considerable evidence implicating the vulnerability of the hippocampus to TBI because of its particular location and structural configuration (Geddes et al., 2003; Grady et al., 2003; Hicks et al., 1993). Although cell counts revealed the significant neuronal loss in the hippocampal CA3 region after purely angular acceleration, neuronal loss was more conspicuous and extensive in the animals subjected to the combined accelerations. In addition to the more prominent direct mechanical injury that occurred after the combined accelerations, as compared with a single type of acceleration, perisomatic axonal damage in the former case might have also contributed to the significant loss of neuronal cells. Neurons would undergo retrograde degeneration because of axonal damage (Buki and Povlishock, 2006; Dusart et al., 2005; Liu et al., 2006). In addition, cell death in the hippocampus also occurred in the animals subjected to the purely linear acceleration, even though the cell counts showed no significant cell loss compared with that seen in the control animals. This result may be relevant to the observed clinical phenomenon that even mild brain injury can lead to persistent post-concussion symptoms (Begaz et al., 2006; Chen et al., 2008).
Disconnection of axons at the time of brain trauma (primary axotomy) is a relatively rare occurrence, with the exception of tissue tearing in the white matter in severe brain injury. Rather, axonal pathology has been shown to develop over the course of hours to days after injury (Adams et al., 1982; Pierce et al., 1998; D.H. Smith et al., 2003). In the current model, EM also identified primary axonal tearing that was induced by the trauma: the axolemma and myelin sheath had broken down in the absence of axonal swelling or terminal bulbs. As mentioned previously, primary axotomy usually occurs in severe DAI (Maxwell et al., 1993; Povlishock et al., 1983). These pathological changes revealed by EM demonstrated further that severe DAI was induced in our experimental model under low loading conditions.
Conclusion
In the current study, we have developed a new experimental model for DAI in the rat, in which pathological and neurological changes are induced that are consistent with the DAI produced in other models and are comparable to the alterations found after clinical DAI. This new experimental model can combine linear and angular accelerations; these two factors usually act together in realistic injury scenarios. Multiple pathological observations showed that DAI can be produced reliably and consistently in our model. Non-linear and synergistic effects caused by the combination of linear and angular accelerations are also revealed in the present study. This mechanism of injury may explain why DAI is a frequent form of TBI, and is paramount for the design of protective measures. Future research will focus on issues such as the characterization of differences in the pathological and functional changes that occur when: the angular acceleration is varied but the linear kinematics remain constant; when the linear acceleration, but not the angular acceleration, is altered; and when both of forms of accelerations are simultaneously changed.
Footnotes
Acknowledgments
We acknowledge Yuying Chen, Zhuying Guo, and Dong Yang for excellent technical support. This work is supported by grants from Shanghai Education Committee (no. 06BZ042), Shanghai Municipal Health Bureau (no. 2008115), and Shanghai Jiao Tong University Medicine and Engineering Cross Fund (no. YG2009MS14).
Author Disclosure Statement
No competing financial interests exist.