
Abstract
Traumatic brain injury (TBI) is the one of the most common forms of head trauma, and it remains a leading cause of death and disability. It is known that the initial mechanical axonal injury triggers a complex cascade of neuroinflammatory and metabolic events, the understanding of which is essential for clinical, translational, and pharmacological research. These can occur even in mild TBI, and are associated with several post-concussion manifestations, including transiently heightened vulnerability to a second insult. Recent studies have challenged the tenet that ischemia is the ultimate modality of tissue damage following TBI, as metabolic dysfunction can develop in the presence of normal perfusion and before intracranial hypertension. In order to elucidate the cellular and molecular changes occurring in TBI as a direct result of neuronal injury and in the absence of ischemic damage, we performed a microarray analysis of expressed genes and molecular interaction pathways for different levels of severity of trauma using an in-vitro model. A stretch injury, equivalent to human diffuse axonal injury, was delivered to rat organotypic hippocampal slice cultures, and mRNA levels following a 10% (mild) and 50% (severe) stretch were compared with controls at 24 h. More genes were differentially expressed following 10% stretch than 50% stretch, indicating the early activation of complex cellular mechanisms. The data revealed remarkable differential gene expression following mTBI, even in the absence of cell damage. Pathway analysis revealed that molecular interactions in both levels of injury were similar, with IL-1β playing a central role. Additional pathways of neurodegeneration involving RhoA (ras homolog gene family, member A) were found in 50% stretch.
Introduction
T
TBI is generally subdivided into mild, moderate, or severe cases, with mild TBI (mTBI) representing the majority of all head injuries (Alexander, 1995; Vos et al., 2002). Different definitions of mTBI exist, but in general it is characterized by a shorter period of reduced consciousness (usually only for seconds to minutes), compared to moderate and severe injuries (Alexander, 1995; Vos et al., 2002). Common complaints of those with mTBI include headache, pain, visual and vestibular disturbances, fatigue, emotional and behavioral deficits, depression, irritability, anxiety, lack of initiative, decreased self-esteem, social difficulties, and personality changes (Comper et al., 2005; Jay et al., 1996).
Mild TBI is difficult to manage properly, as those who do present for medical care are rarely admitted. Those who attend accident and emergency are often not seen by a specialist, and in many cases receive no specific advice or care at all (Comper et al., 2005; Jay et al., 1996; Teasdale and Jennett, 1974; Vagnozzi et al., 2007).
It is clear that the response of the brain to trauma is complex and involves many biochemical and molecular changes. Further, the molecular processes triggered by mTBI, in which little if any discernable cell death occurs, are likely to be, at least in part, different from those occurring after more severe injury.
Numerous researchers have examined the expression of individual genes whose products contribute to recovery or impairment of function following head injury. However as yet, a global analysis of messenger RNA (mRNA) expression in reliable models of TBI has not been carried out. The main drawback of the analysis of individual genes is that the relationships between the different pathways are extremely complex and highly variable, not only with respect to severity of injury, but also over time. An ideal approach would be to look simultaneously at a transcriptional profile at defined time points, and in response to different levels of severity of insult.
In this study, microarray technology was utilized to examine gene expression profile in an in-vitro model of rat hippocampal slice culture stretched at specific Lagrangian strains (10% and 50%), through the dynamic deformation of the silicone membrane to which the slices were adhered. This produces a strain field in the tissue that mimics the in-vivo tissue deformation experienced by brain tissue during acceleration/deceleration trauma.
This model has been amply validated and allows the study of the cellular effects of TBI without confounding systemic variables, such as hypoxia, hypotension, or sepsis. Although the latter are all powerful determinants of outcome in clinical practice, the identification of the molecular mechanisms involved in the pathophysiology of TBI is a critical step in the identification of reliable clinical biomarkers of central nervous system (CNS) injury, drug development, and stem-cell research.
Methods
Organotypic hippocampal slice cultures
Organotypic hippocampal slice cultures maintain their anatomical organization of neurons and glia, and thus provide an ideal model for studying the effects of stretch-induced brain trauma (Kumaria and Tolias, 2008).
The hippocampal cell cultures were prepared using a method initially described by B. Morrison (Morrison et al., 2006), and all procedures used were in accordance with U.K. regulations under the Animals (Scientific Procedures) Act of 1986.
Briefly, the septal portion of hippocampi from 8- to 10-day-old Wistar rats were removed and sliced using a McIlwain tissue chopper (Harvard Apparatus, Edenbridge, U.K.) to a thickness of 400 μm. These were then placed in ice-cold Geys solution supplemented with 5 mg/mL
Prior to preparation of the slices, the wells were assembled by attaching the non-porous silicone membranes (0.25 mm; Specialty Manufacturing, Saginaw, MI) to custom-made stainless steel wells. This assembly was placed in a glass Petri dish and sterilized. Also, 48 h prior to hippocampal tissue culturing, the silicone membranes were pretreated with a coating solution of 320 μg/mL poly-
After 2 days in vitro, the neurobasal medium bathing the slices was replaced with full serum-containing medium, consisting of 25% heat-inactivated horse serum, 25% Hanks' balanced salt solution, 50% minimum essential medium (all Invitrogen), 1 mmol/L glutamine (Sigma), and 5 mg/mL
Five cultures (each containing 4 slices from the same rat) were used for each experimental group.
Stretch injury device
The biomechanics of the stretch injury device have been described in detail previously (Morrison et al., 2006). Briefly, however, it is designed to stretch the organotypic hippocampal slice cultures through dynamic deformation of the silicone membrane upon which the tissue slices were grown and adhered. The strain field is produced by a mechanical loading mechanism that mimics the in-vivo tissue deformation thought to be experienced during TBI.
The stainless steel well and silicone membrane were held in place by a circular clamp on a heated platform (37°C). The membrane was then displaced over a fixed, hollow indenter to generate an equi-biaxial strain field, subjecting the cultures to a single stretch injury at a specified Lagrangian strain and constant strain rate. The strain was controlled by a linear actuator (BEI Kimco, Vista, CA), and maintained under feedback control through a linear encoder (Renishaw, New Mills, Wotton-under-Edge, U.K.), and motion control board (Precision Motion Dynamics, Victoria, Canada).
Stretching the organotypic hippocampal slice cultures
After approximately 10 days in vitro, the viability of the organotypic hippocampal slice cultures was assessed by viewing under a light microscope to ensure the slices were adhered to the membrane, and to see if clear definition of the neuronal regions (CA1, CA3, and dentate gyrus) could be seen. Any cultures that were not completely adhered to the silicone membrane were discarded.
The cultures were placed in fresh, serum-containing full medium with 5 μg/mL of propidium iodide (PI) (Molecular Probes, Paisley, U.K.), an exclusion dye, for 2 h, and then they were imaged using a Leica DM-IRBE epifluorescence microscope (Leica Microsystems Ltd., Milton, Keynes, U.K.), and any cultures expressing excessive PI fluorescence were discarded.
Images were acquired using a 5 × NA 0.12 lens and a cooled Hamamatsu camera and analyzed with Volocity (Improvision, Coventry, U.K.) 3-D imaging software.
The medium was removed from the healthy cultures and the wells were clamped to the stretch injury device. The membrane was then displaced to generate a strain field, and the cultures were subjected to a single stretch injury at a Lagrangian strain of 10% or 50%, at a constant strain rate of 20 sec–1, or in the case of controls, no stretch at all.
Propidium iodide fluorescence
Following the stretch insult, the cultures were placed in fresh full serum-containing medium. Twenty-four hours post-injury, the organotypic hippocampal slice cultures were examined under the epifluorescence microscope to assess the percentage of cell death. Neuronal damage was expressed as a percentage of the area in which PI fluorescence was detected above threshold within a cell layer divided by the total area of that cell.
The mean percentage area of PI fluorescence for controls, 10% stretch, and 50% stretch were analyzed and compared using one-way independent group analysis of variance (ANOVA). Following one-way ANOVA, a post-hoc multiple comparison procedure was performed to compare pairs of group means. As one group represented control data, Dunnett's test was performed to compare 10% and 50% stretch cell damage to controls, but not to each other. All tests were two-tailed, and statistical significance was set at p ≤ 0.05.
cDNA microarray
Total RNA was extracted from tissue cultures in a monophasic solution of phenol and guanidine isothiocyanate (Trizol; Invitrogen) as previous described (Vagnozzi et al., 2007).
The integrity and concentration of the total RNA were determined using the RNA 6000 Nano Assay Kit and a Bioanalyzer 2100 according to the manufacturer's protocols (Agilent Technologies, Stockport, U.K.).
The labeling of complimentary RNA (cRNA) with the fluorophores cyanine-3 (cy-3) and cyanine-5 (cy-5) (PerkinElmer/NEN Life Sciences, Milan, Italy) and its subsequent amplification was completed using the Low RNA Input Linear Amplification Kit (Agilent) according to the manufacturer's protocol.
Briefly, in this procedure one sample (control) is labeled with cy-3 (which was excited by a 532-nm laser), and one sample (either 10% stretch or 50% stretch) with cy-5 (which was excited by a 633-nm laser). A primer, which contains poly (oligo) dT and a T7 polymerase promoter, was annealed to the sample RNA. Next, using the T7 RNA polymerase, cRNA was synthesized and amplified, while simultaneously cy-3- or cy-5-labeled CTP is incorporated into it.
The cRNA was assessed using a NanoDrop® ND-3300 Fluorospectrometer (Agilent) to ensure that sufficient cRNA of appropriate quality had been prepared.
Once labeling was complete, equal amounts of cy-3- and cy-5-labeled probes were simultaneously applied to a 4 × 44 K whole rat genome microarray (Design 14879; Agilent) for a competitive hybridization reaction using a gene expression hybridization kit (Agilent), according to the manufacturer's protocol.
Following hybridization, the array was washed using the gene expression wash buffer kit (Agilent), dried, and then scanned using a DNA microarray scanner (Agilent), and the data were processed using feature extraction software (Agilent).
Data analysis
The datasets, pre-processed by Agilent Feature Extraction software, were analyzed by Genespring GX 10.0.2. Automatic flags applied by the Feature Extraction software were used to localize and exclude from the analysis any probes that were non-uniform, population outliers, saturated, absent, not positive and significant, or not above background.
The signal from each spot was calculated as the average intensity minus the average local background. Expression ratios of cy-5:cy-3 were normalized using LOESS, a method that takes into account and corrects for intensity-dependent artifacts in the measurements.
The mean log2 ratios for each probe were found across the two replicates, and the probes that revealed a >1.5-fold difference in expression (higher or lower) compared to the control sample were identified.
The gene ontology (GO) term analysis was completed using a p-value of <0.1, with Benjamini-Yekutieli false discovery rate correction.
The list of differentially expressed genes were analyzed for any gene ontologies that were statistically over-represented and assigned to functional categories (GO terms) using an enrichment analysis.
Using the GO terms and lists of differentially expressed genes produced following enrichment analysis, the European Bioinformatics Institute's (EBI) IntAct database was used to analyze molecular interaction data and generate a pathway of these molecular interactions and reactions for both 10% and 50% stretch.
Gene expression data have been deposited in ArrayExpress EBI (
Real time-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was used to validate the expression of several genes shown in the microarray analysis.
First, the reverse transcription of total RNA was performed for control, 10% stretch, and 50% stretch samples. From each sample, 1 μg of total RNA, 500 ng of oligo dT primers (Roche Molecular Biochemicals, Lewes, Sussex, U.K.), and 200 U of Superscript II Reverse Transcriptase in a total volume of 20 μL of 1× First Strand Buffer (Invitrogen) were incubated at 42°C for 60 min.
The concentration and purity of the resulting cDNA was determined with a ND-1000 UV-Vis Spectrophotometer (NanoDrop, Labtech Internet NAL, East Sussex, U.K.).
RT-qPCR was carried out on the single-stranded cDNA yielded from the reverse transcription using a one-step PCR kit, Rat Custom real-time PCR assay for use with SYBRgreen chemistry (PrimerDesign Ltd., Southampton, U.K.), and a Rotor-Gene 6000 real-time thermocycler (Corbett Robotics Ltd., Corbett Research, Cambridge, U.K.) according to the manufacturer's protocol, for 40 cycles.
The genes selected for RT-qPCR and sequences of primers were: S100a6 (S100 calcium binding protein A6) Rattus norvegicus (NM_053485), forward GCACACCCTGAGCAAGAAG, reverse CCTTGTTACGGTCCAGATCATC; and interleukin-1β (IL-1β) Rattus norvegicus (NM_031512), forward AGCACCTTCTTTTCCTTCATCTT, reverse CAGACAGCACGAGGCATTTT. These primers were designed by Primer Design Ltd. (Southampton, U.K.).
For accurate gene expression measurements with RT-qPCR, it is essential to normalize results to a fixed reference, and therefore the constitutively expressed housekeeping gene B2M (beta-2-microglobulin) Rattus norvegicus (NM_012512) was selected using the geNorm Housekeeping Gene Selection Kit (Primer Design Ltd.) from a list containing 12 candidate reference genes.
Results
PI fluorescence
Stretching the organotypic slice cultures resulted in cell damage, as measured by uptake of the fluorescent exclusion dye PI. Representative fluorescence images of the slice cultures were taken at 24 h after 10% stretch, 50% stretch, and of control. The percentage area of red fluorescence compared to the total area of the hippocampal slice of each image was later analyzed and compared. Analysis of percentage cell damage can be seen in Figure 1.

Cell damage in control, 10% stretch, and 50% stretch cultures. Damage within the CA1 and CA3 regions as determined by propidium iodide fluorescence at 24 h post-injury is significantly increased following a 50% stretch injury compared to controls. No significant increase in cell damage occurred following 10% stretch compared with controls (p = 0.44). Data are presented as mean ± standard deviation (**p < 0.001 versus controls).
One-way ANOVA showed statistically significantly different cell damage between experimental sets (p < 0.0001). When comparing the two stretch groups with controls with Dunnett's test, the 10% stretch injury slices did not show a significantly different percentage of cell death (mean cell death 3.07%, SD ± 1.91 in 10% stretch, versus mean cell death 0.31% SD ± 0.38 in controls; p = 0.44), as expected in mild TBI, thus confirming the validity of this model. The damage occurring following 50% stretch, however, was significantly greater (mean 42.23%, SD ± 13.17; p < 0.001), again confirming this to be a valid model for this severity of injury.
cDNA microarray
Traumatic brain injury activates complex cascades of biochemical changes, and these changes alter the expression of genes, whose products contribute to the pathophysiology seen following brain trauma.
Of more than 41,000 gene probes on the microarray, 18,459 met the inclusion criteria for detectable expression.
In the 10% stretch sample, a total of 999 probes were differentially expressed, and for 50% stretch, 587 probes were differentially expressed, either increased or decreased, compared to controls, by a minimum fold change of 1.5.
After data normalization and fold change calculation, a comparison was made between genes differentially expressed in 10% stretch and 50% stretch compared with controls.
We found that in 10% stretch 210 genes were upregulated, in 50% stretch 161 genes were upregulated, and of these, 30 genes were increased in both experimental groups.
A comparison between differentially downregulated genes showed in 10% stretch 789 genes were downregulated, and in 50% stretch 426 genes were downregulated, and of these, 307 genes were decreased in both groups (Fig. 2).
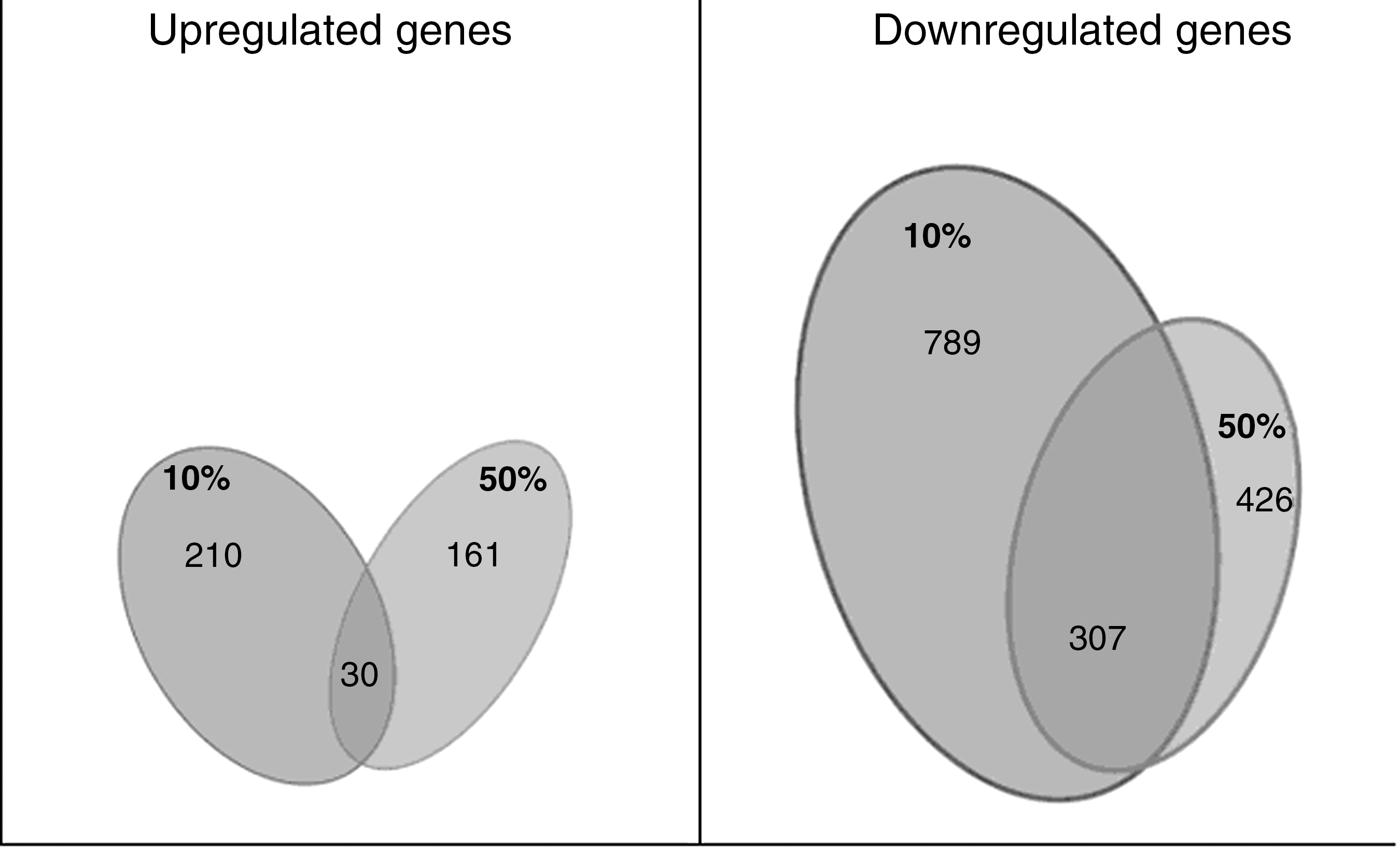
Gene expression following 10% and 50% stretch injury. Venn diagram illustrating differentially expressed genes at the two different injury intensities. Overlapping areas show genes for which expression was altered in both levels of stretch injury.
The full dataset of all differentially expressed is available as a supplementary file (Supplementary Table 1) (see online supplementary material at
Gene analysis and ontology
The differentially expressed genes were assigned to functional categories using gene enrichment analysis. This process evaluates microarray data and provides categories based on genes sharing the same GO term, as defined in published information on biochemical pathways in previous experiments (Kerrien et al., 2007; Subramanian et al., 2005).
The altered genes in 10% stretched cells can be broadly classified into nine functional groups: chromatin assembly or disassembly, nucleosome organization, organ development, chromatin assembly, developmental process, nucleosome assembly, multicellular organismal development, system development, and anatomical structure development. All of these were found to be within the GO “Biological Process” (Fig. 3).

Gene ontology (GO) following 10% stretch injury. Diagrammatic representation of the functional categories (GO terms) associated with differentially expressed genes following a mild (10%) stretch injury. All share the same gene ontology: Biological Process.
The genes found to be differentially expressed following a 50% stretch were in three different GOs: “Molecular Function,” “Cellular Component,” and “Biological Process,” and classified within 13 functional groups. In Molecular Function the categories were G-protein coupled receptor binding, receptor binding, cytokine activity, binding, chemokine activity, and chemokine receptor binding. The categories defense response, inflammatory response, behavior, response to external stimulus, response to wounding, and neutrophil chemotaxis were found to be in Biological Process. The GO Cellular Component was represented by one functional category, extracellular region (Fig. 4).
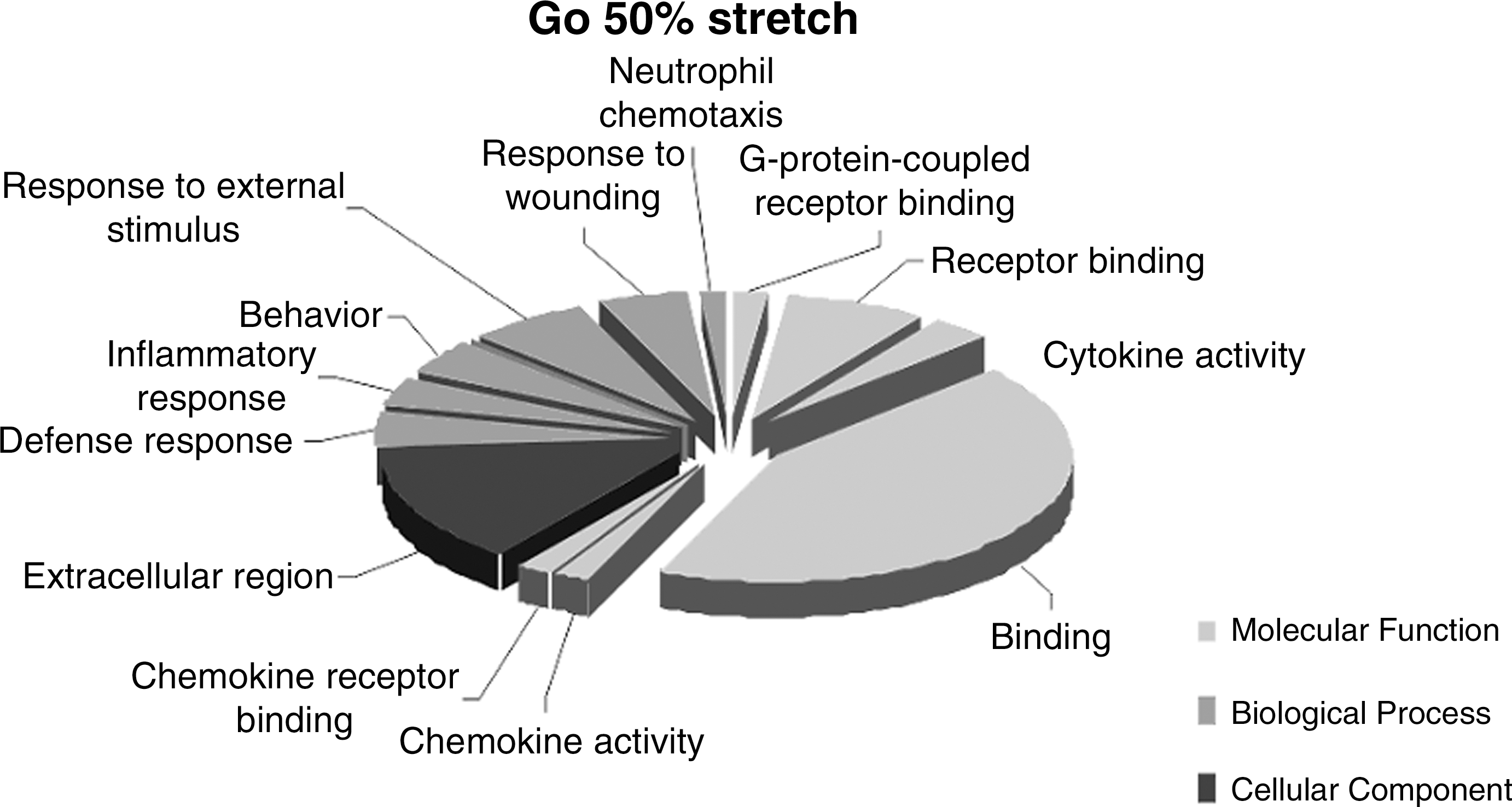
Gene ontology (GO) following 50% stretch injury. Diagrammatic representation of the functional categories (GO terms) associated with differentially expressed genes following a severe (50%) stretch injury. Three gene ontologies were found to correspond with these genes: Biological Process, Molecular Function, and Cellular Component.
Pathway analysis
Using the GO terms produced following gene enrichment, the EBI IntAct database was used to analyze molecular interaction data and generate a pathway of these molecular interactions and reactions for both 10% and 50% stretch. The pathways produced are the extensive interactive diagrams shown in Figure 5 (for 10% stretch) and Figure 6 (for 50% stretch).
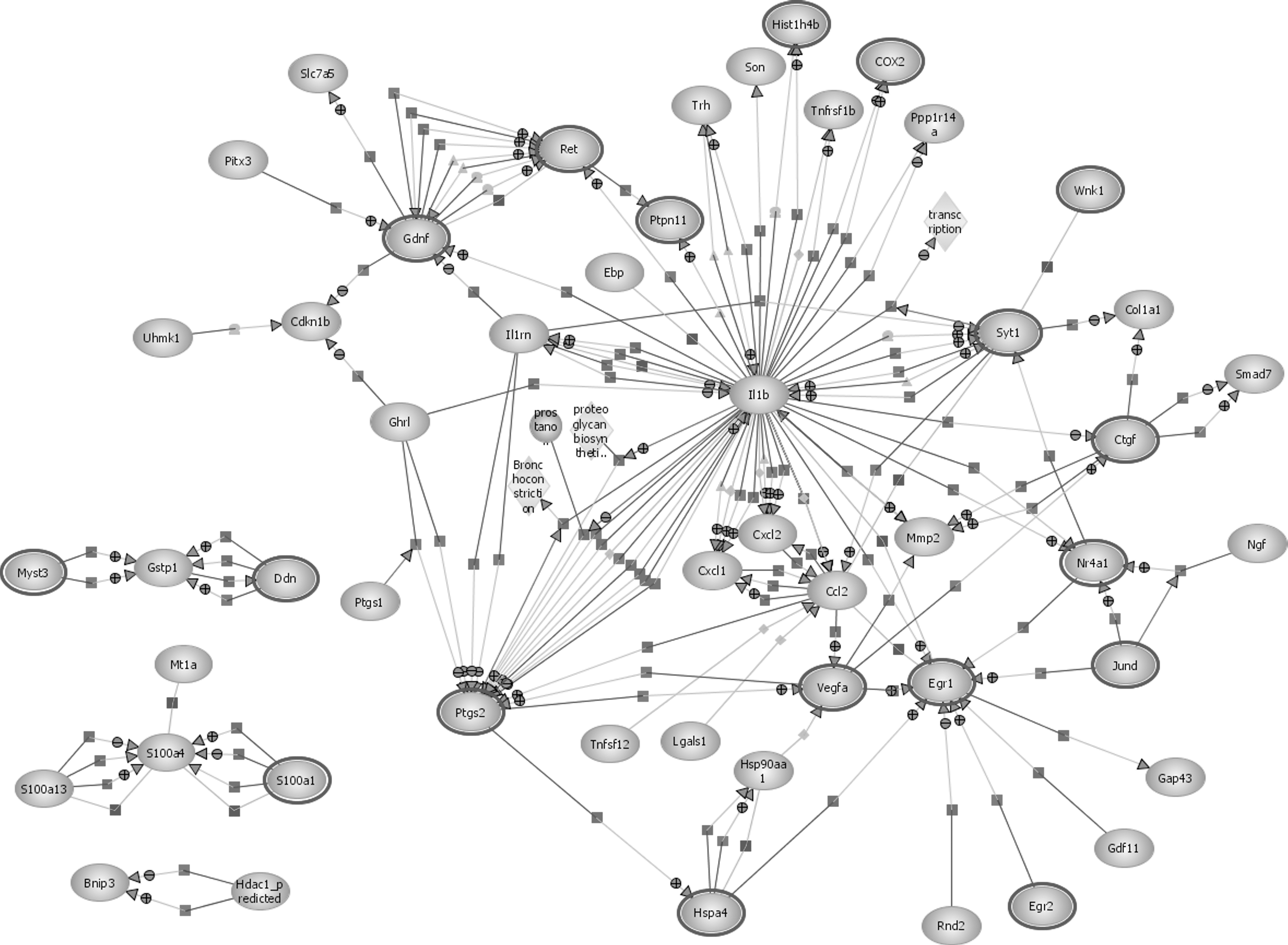
Pathway analysis following 10% stretch injury. Pathway analysis diagram showing interactions between various genes with differential expression for a mild traumatic brain injury (10% stretch).
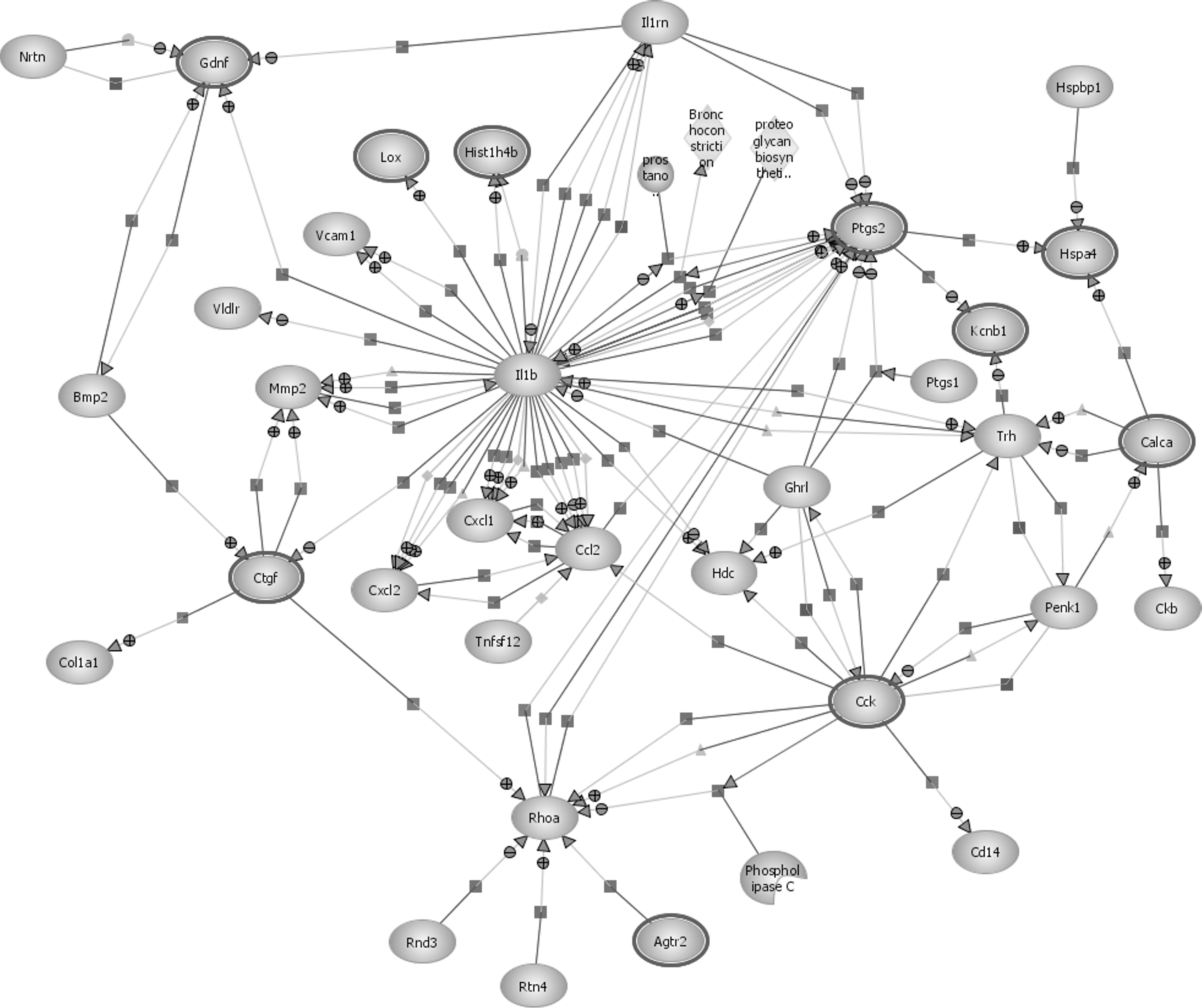
Pathway analysis following 50% stretch injury. Pathway analysis diagram showing interactions between various genes with differential expression for severe traumatic brain injury (50% stretch).
The pathway analysis for both a 10% and 50% stretch injury reveals that the pathways share many interactions, while also highlighting key differences. Proinflammatory cytokines appear to have a central role in molecular interactions following both a mild and a severe brain injury. In both pathways it can be clearly seen that IL-1β plays an important role and has numerous interactions with other genes. Prostaglandin-endoperoxide synthase 2 (PTGS2), also known as cyclooxygenase-2 (COX-2), also appears to be vital in both 10% and 50% stretch, with many regulation, expression, and metabolism interactions with IL-1β.
A crucial pathway that became evident following 50% stretch, but that was not present in the mild injury pathway analysis, involved the myelin growth inhibitor rtn4 (Nogo-A), targeting and positively regulating the GTPase RhoA (ras homolog gene family, member A), which is implicated in the neurodegenerative process.
Both pathway analysis diagrams are available as supplementary clickable files (Supplementary Figures 1 and 2) (see online supplementary material at
RT-qPCR analysis
In order to validate the expression profiles observed, RT-qPCR analysis was performed by selecting two genes with either a high or low fold change, as seen in cDNA microarray analysis results for both 10% and 50% stretch samples.
The RT-qPCR analysis confirmed the upregulation of IL-1β parallel to that seen in both 10% and 50% stretch injury following microarray analysis, and also confirmed the downregulation of the gene S100a6, also in parallel to the microarray analysis (Fig. 7A and B).

RT-qPCR analysis of (
Discussion
The results of this investigation show that the pathophysiology following TBI is complex and involves alterations in the expression of genes, whose products are involved in a variety of cellular processes that contribute to the behavioral deficits and pathological consequences of brain injury.
The identification of genes induced in response to brain injury is currently an area of intense research, particularly since the development of the microarray. The ability to use probe sequences that are complimentary to thousands of genes has allowed the study of large-scale mRNA changes associated with a variety of physiological and pathological processes (Luo and Greshwind, 2001).
Using cDNA microarray technology, we report that following stretch injury to hippocampal slice cultures, the expression of 999 genes in 10% stretch and 587 genes in 50% stretch were altered, compared with controls.
The altered genes in 10% stretched cells clustered in the “Biological Process” group, which has shown to be involved in the structural damage of cellular architecture.
Most of the genes expressed following 10% stretch are involved in signal transducer activity, regulation of transcription, and cell communication. This indicates that even after a mild injury of 10% stretch, intense activity involving transcription and signaling exchange is initiated.
Additionally, we have found that following 10% stretch, certain genes involved in the apoptotic process, such as Vdac1 (voltage-dependent anion-selective channel protein 1), Sh3glb1 (SH3-domain GRB2-like endophilin B1), Phlda1 (leckstrin homology-like domain, family A, member 1), Rock1 (Rho-associated coiled-coil containing protein kinase 1), and Eif4g2-predicted (eukaryotic translation initiation factor 4 gamma, 2) were downregulated. Further, an upregulation was seen in genes involved in the anti-apoptotic process, such as Ccl2 (chemokine [C-C motif] ligand 2), Vegfa (vascular endothelial growth factor A), BIRC3 (baculoviral IAP repeat-containing 3), Tsc22d3 (TSC22 domain family, member 3), Bnip3 (BCL2/adenovirus E1B 19-kDa interacting protein 3), and Nr4a1 (nuclear receptor subfamily 4, group A, member 1).
The majority of these expression changes were only found following 10% stretch, indicating that these hippocampal cell cultures have activated protective and repair mechanisms.
Following a severe 50% stretch, the genes were clustered in three different GOs: “Molecular Function,” “Cellular Component,” and “Biological Process,” and the majority of genes seen to be upregulated are involved in inflammatory events, apoptosis, and necrosis, such as Casp1 (caspase 1, apoptosis-related cysteine peptidase), Nlrp3 (NLR family, pyrin domain containing 3), and as can clearly be seen in the pathway analysis, Rtn4 (reticulon 4) and RhoA.
As expected following a severe traumatic injury, in addition to the typical increase in the inflammatory process and the activation of apoptotic and necrotic events, through GO analysis we have revealed the upregulation of genes involved in the activation of oxidative and nitrosative stress, as Sod2 (superoxide dismutase 2), Rasd1 (RAS, dexamethasone-induced 1), and COX-2.
In both injury groups, the majority of genes were downregulated. Following a 10% stretch, 789 genes were downregulated, and after 50% stretch, considerably fewer, 426 genes were downregulated. Studies of hippocampal gene expression following preconditioning and tolerance to epilepsy and ischemia have also shown a similar global downregulation of gene expression following the initial mild preconditioning stimuli. In these injuries, it is thought that gene suppression is a mechanism of the reprogramming response thought to occur in tolerance (Jimenez-Mateos et al., 2008).
The suppressed expression of genes appears to be a significant feature of reprogramming that occurs after other types of CNS injury, and may explain our results following mild TBI. The mechanism of reprogramming leading toward gene suppression is largely unknown, but it appears to be a function of the preconditioning stimuli, and may involve increased transcriptional silencing (Roopra et al., 2001) or other post-transcriptional mechanisms (Saugstad et al., 2007).
Our data globally showing the large number of downregulated genes following both injury levels, and especially the increase in the number of genes differentially expressed following 10% stretch and 50% stretch, allows us to conclude that the two-dimensional stretch of organotypic hippocampal slice culture model appears comparable to mild TBI in vivo.
Having shown that more genes are differentially expressed following a mild brain injury than a severe injury further supports the notion that even following a mild TBI, in which an absence of radiological and clinical abnormalities is the norm, an invisible complex cellular response is initiated and distinct neuronal dysfunction occurs. This corroborates previous data obtained after in-vivo TBI in a rodent model (Tavazzi et al., 2007; Vagnozzi et al., 2007), but it also confirms that these are primary cellular effects that are not determined by local blood or oxygen delivery, or by systemic factors. This challenges a widely-accepted pathophysiological model of TBI that has cerebral swelling and tissue ischemia at its center. In this model, cerebral swelling caused by trauma causes a rise in intracranial pressure (ICP), which consequently produces a drop in cerebral perfusion pressure (CPP; CPP = mean arterial pressure – ICP). ICP control remains the mainstay of modern TBI management in both American (Brain Trauma Foundation:
In this work we also showed that cytokines play a central role in the activation of different pathways following injury, as has been demonstrated in previous studies (Ghirnikar et al., 1998; Pinteaux et al., 2009). It is thought that these messengers are involved in the communication between injury-activated astrocytes and microglial cells, altering the glial response to injury (Rothwell and Luheshi, 2000; Saavedra et al., 2007). IL-1 is one of the most widely studied proinflammatory cytokines, and it has been implicated in a number of neurodegenerative disorders, both acute and chronic (Allan et al., 2005; Lucas et al., 2006). IL-1β may also to lead to the activation of Rho (as shown in the 50% stretched cells), an intracellular GTPase regulating the neuronal response to myelin growth inhibitory proteins and the regeneration of axons, and associated with an increased susceptibility to apoptosis (Domeniconi and Filbin, 2005; Dubreuil et al., 2006).
Myelin contains three major growth inhibitors: Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein, that impede regeneration of axons in the CNS (Domeniconi and Filbin, 2005). Findings suggest that these molecules share the same receptor complex between p75NTR (p75 neurotrophin receptor) and the Nogo receptor, which in turn signals through RhoA activation and induces growth cone collapse (Dubreuil et al., 2006).
Through the molecular interaction pathway analysis performed in this study, we have shown that initiation of inflammation is a key process following both mild and severe TBI. In agreement with previous reports (Fan et al., 1995; Strauss, 2008), the microarray analysis revealed that the transcription levels of the proinflammatory cytokine IL-1β and the inducible form of COX-2 were significantly elevated at 24 h post-injury. COX-2 is crucial in the progression of secondary brain injury, increasing total brain prostaglandin and reactive oxygen species (ROS) levels (Strauss, 2008). It is mainly through the production of superoxide anion products that it is thought that COX-2 expression contributes to excitotoxic cell death (Strauss, 2008), and together with the damage done by other ROS to lipids, protein, and DNA, the excitotoxic process is exacerbated (Strauss, 2008).
Subsequent to the initial mechanical insult, the inflammatory response is an important mechanism of secondary damage, contributing to cell death and necrosis (Dubreuil et al., 2006; Lenzlinger et al., 2001). The immune reaction seen after TBI is characterized by the activation of microglia cells, rapidly followed by the recruitment of macrophages to the site of injury (Dubreuil et al., 2006; Lenzlinger et al., 2001). The invasion of macrophages further augments this inflammatory cascade by inducing proinflammatory cytokines, such as tumor necrosis factor-α and IL-1β, which are known to lead to the activation of Rho (Berry, 1982; Neumann et al., 2002). We have demonstrated that this effect on Rho occurs in our in-vitro model of TBI, but specifically only after a severe 50% stretch injury. In fact, the main difference observed between the 10% and 50% injury concerns the implication of RhoA and its role in the neurodegenerative process.
Following TBI in humans, it has been shown that the GTPase RhoA is upregulated, with a specific increased expression in reactive glia and swollen neuritis (Brabeck et al., 2004; Dubreuil et al., 2006). Growth inhibitors released from damaged white matter have been shown to act through the RhoA pathway and to contribute to widespread axonal injury, tissue damage, and a lack of regeneration following TBI (Dubreuil et al., 2006; Maxwell et al., 1997).
Of the three major growth inhibitory proteins found within myelin, MAG and Rtn4 (also known as Nogo) were found to be upregulated following 50% stretch. These proteins are known to block axon regeneration by acting through the RhoA pathway (Domeniconi and Filbin, 2005; Dubreuil et al., 2006). The activation of this gene may indicate the transition point from a mild reversible injury to a more severe and permanent one. This view is supported by the results from the PI analysis we performed 24 h after injury, in which a significant increase in cell damage and death was seen following a severe injury compared with a mild injury and controls. This may also provide a focal point for therapeutic intervention.
Following mTBI, the genes differentially expressed, seen both in this study and in previous investigations, have functionally been implicated in regeneration, reorganization, and plasticity following injury. This suggests that the modulation seen in the expression of these genes may enhance recovery and/or halt neurological dysfunction following mild injury. Conversely, after a severe TBI, the subsequent gene expression leads to irreversible damage and cellular death.
The understanding of these mechanisms may hold the key to future neuroprotective, repair, and neuroregeneration research.
Footnotes
Acknowledgments
We wish to thank the Wessex Neurological Centre Trust for funding this study and for their continuing support for our research. We also wish to thank the Italian Ministry of University and Scientific Research for funding Dr. Tavazzi's work (research grant PRIN 2007JBHZ5F-COFIN 2007). We are also grateful to Prof. Mark Bradley of the School of Chemistry, University of Edinburgh, for his support.
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.