
Abstract
Little is known about the molecular events following severe traumatic brain injury (TBI) in humans and to date there are no efficient therapies for the treatment of patients. In this study, the first of its kind in human tissue, a total of 21 post mortem trauma brain samples were analyzed. The inflammatory response within the brain tissue was explored by measuring the expression of various inflammatory cytokines at the mRNA and protein levels. These mediators were interleukin (IL)-1β, IL-2, IL-4, IL-6, IL-8, IL-10, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF). This study shows for the first time in human brain tissue that 1) pro-inflammatory mediator protein levels are significantly increased in situ following acute brain injury while anti-inflammatory cytokines protein levels remain unchanged; 2) the cerebral inflammatory response begins within minutes of acute TBI, much earlier than previously thought; 3) IL-6, IL-8, TNF-α, and IL-1β mRNA levels are significantly increased following injury; 4) the rise in cytokine protein level coincides with increased levels of their mRNAs suggesting that traumatic injury elicits an immediate cerebral inflammatory response. Altogether these data confirm and extend previous observations on the release of cytokines in the cerebrospinal fluid of severe TBI patients. Finally, this study highlights the need to characterize the cell source of cytokines and elucidate their mode of action.
Introduction
T
Primary brain damage occurs immediately at the time of impact and involves mechanical cell destruction. Secondary brain damage develops within minutes to months after the injury and is mediated by multiple physiological and molecular cascades leading to ongoing neuronal degeneration (Morganti-Kossmann et al., 2009). Inflammatory mediators, excitatory amino acids, and free radicals have been shown to play key roles in the exacerbation of neural injury following TBI through the initiation of various neurotoxic pathways, contributing to cell membrane breakdown and cell death (Werner and Engelhard, 2007). In addition, a recent study on 8000 TBI patients showed that imbalance in systemic physiological conditions like hypertension and hypoxia are linked with the worst outcome in severe diffuse brain injury (Maas et al., 2007).
The long-established approach in the development of efficacious therapeutics in humans and animal models of TBI aims at inhibiting the molecular pathways leading to secondary brain damage. The inflammatory response is an important component of TBI and is characterized by the release of pro- and anti-inflammatory mediators with dual and opposing roles: on the one hand enhancing brain damage through the release of neurotoxic substances, but on the other hand promoting repair of the injured tissue by stimulating the synthesis of neurotrophins (Correale and Villa, 2004; Lucas et al., 2006; Morganti-Kossmann et al., 2001). This harmful–beneficial dual aspect makes the neuroinflammatory cascade a challenging therapeutic target and more research specifically in humans is needed since most of the data collected in this field are derived from studies on animal models of TBI (Cernak, 2005; Csuka et al., 2000; Hans et al., 1999; Rancan et al., 2001; Stahel et al., 2000).
Several approaches have been used over the years to monitor and elucidate the role of neuroinflammation in TBI patients. The first and more accessible approach involved the analysis of cytokines in cerebrospinal fluid (CSF), which is thought to mirror brain production. Our group was one of the first to detect raised cytokine concentrations in the CSF of TBI patients up to 3 weeks post-injury (Csuka et al., 1999; Hayakata et al., 2004; Kossmann et al., 1996, 1997; Morganti-Kossmann et al., 1999, 2001; Pleines et al., 1998; Whalen et al., 2000). A second experimental approach developed in recent years in the clinic relies on the insertion of a microdialysis catheter in the frontal cerebral cortex of TBI patients (Hillman et al., 2005; Hutchinson et al., 2007; Mellergard et al., 2008; Winter et al., 2004). Despite the controversies surrounding the use of a somewhat invasive procedure, which may itself cause damage, this technique allows for in situ measurement of cytokines produced by the brain parenchyma. The third and last approach used so far in humans is the examination of contused brain biopsies obtained from patients undergoing surgery to evacuate the injured tissue (Holmin and Höjeberg, 2004; Stefini et al., 2008). Even though this approach provides valuable information at the tissue level, it presents few technical limitations (low efficiency of cytokine retrieval), which may not reflect accurately the events occurring in the whole injured brain.
Despite the large number of studies examining cytokine expression following TBI, data on human brain tissue is almost nonexistent due to the lack of tissue available for research. Therefore in the present study we used post mortem brains of patients who died following acute closed head injury to determine for the first time the protein concentration and mRNA expression level of several inflammatory mediators, namely, interleukin (IL)-1β, IL-2, IL-4, IL-6, IL-8 (also known as CXCL8), IL-10, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF). We also examined the morphological changes of the injured brain by assessing the extent of astrocyte and microglia activation as well as the axonal accumulation of amyloid precursor protein (APP) and neurofilament (NF) to detect axonal damage. Finally, we correlated the degree of axonal damage with the cellular inflammation in post mortem TBI tissue.
Methods
Human post mortem brain tissue
All procedures were conducted in accordance with the Australian National Health and Medical Research Council's National Statement on Ethical Conduct in Human Research (2007), the Victorian Human Tissue Act 1982, the National Code of Ethical Autopsy Practice, and the Victorian Government Policies and Practices in Relation to Post Mortem.
Trauma brain samples from 21 individuals who died after closed head injury were obtained from the Australian Neurotrauma Tissue and Fluid Bank. Cases were aged between 18 and 78 years (mean 47 years). The causes of injury included motor vehicle accident, motorbike accident, nursing home accident, household accident, stair accident, and falls. The post mortem intervals varied between 40 and 129 h (mean 82 h). In order to compare changes in the inflammatory response between the acute and delayed time after injury, patients were grouped as follows: 14 cases (12 men and 2 women) had a survival time of less than 17 minutes, designated “early” group, when death occurred upon arrivals of the paramedics (Table 1, cases 1–14), while 7 cases (5 men and 2 women) were selected with a survival time between 6 and 122 h (mean 40 h) and were designated “late” group (Table 1, cases 15–21). For both groups, the brain region analyzed was located in proximity of the injured tissue and was identified macroscopically by a neuropathologist (CM). A second brain region also analyzed for the late group was located in the contralateral brain hemisphere where no macroscopic damage was detectable. Control brain samples of 13 individuals, aged between 16 and 78 (mean 58 years), without brain injury or other neuropathologies were obtained from the National Neural Tissue Resource Centre of Australia. Clinical information and epidemiological details of all patients are described in Table 1.
Cases 1–14: early cases with a survival time between 0 and 17 min; Cases 15–21: late cases with a survival time over 6 h; Cases 22–34: control cases. All brains were obtained at autopsy.
PMI, post mortem interval (time between death and brain retrieval); M, male; F, female.
Cytokine protein analysis
Fresh frozen brain cortex samples (100 mg) were homogenized using an ultra-turrax (Ika, Wilmington, NC) in 400 μL of the following buffer: Tris 5 mM, NaCl 15 mM, 1% Triton X-100. Homogenates were placed on ice and on a shaker for 90 min then centrifuged at 2000 g for 10 min at 4°C. The supernatants were stored at −80°C until analysis. Total protein concentrations were determined using the Bradford method (Bio-Rad, Hercules, CA) according to the manufacturer's instructions.
The production of eight cytokines in brain homogenates was determined using the Bio-Plex cytokine assay (Bio-Rad), a multiplex bead-based assay designed to quantitate multiple cytokines in a single sample, following the manufacturer's instructions and as shown previously by our group for mouse brain (Bye et al., 2007). The Human 8-plex A panel used includes IL-2, IL-4, IL-6, IL-8, IL-10, GM-CSF, IFN-γ, and TNF-α, while the levels of IL-1β were measured using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Cytokine mRNA analysis
Total RNA was extracted from fresh frozen brain cortex tissue (100 mg) using TRIzol Plus RNA purification kit and following to the manufacturer's instructions (Invitrogen, Carlsbad, CA). The concentration and purity of the RNA samples were assessed using the Nanodrop1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) while the RNA integrity was assessed using the Agilent 2100 bioanalyzer (Agilent Technologies, Waldbronn, Germany). Prior to using these samples further, a minimum ratio of absorbance at 260 and 280 nm of 2 was considered as pure RNA and a minimum RIN (RNA integrity number) value of 6 was established as a good quality RNA.
The total RNA fractions of each sample were then converted to cDNA using SuperScript III reverse transcriptase (Invitrogen) and oligo d(T)20 as primer, and taken as template for real-time quantitative polymerase chain reaction (Q-PCR) to compare gene expression following brain injury. Q-PCRs were carried out using TaqMan Universal master mix (Applied Biosystems, Foster City, CA) and the 7900HT Fast Real-Time PCR system (Applied Biosystems). TaqMan gene expression assays for IL-6 (Hs00174131_m1), IL-1β (Hs00174097_m1), TNF-α (Hs00174128_m1), and IL-8 (Hs01567912_g1) were used in our study (Applied Biosystems). The relative quantitation was achieved by applying the comparative CT method (ΔΔCT) whereby the mRNA levels were normalized against the level of the 18S ribosomal RNA mRNA (TaqMan gene expression assay Hs99999901_s1) and the control group was used as the calibrator.
Immunohistochemistry
Immunohistochemistry was performed on 7-μm paraffin-embedded cortex and corpus callosum sections using the following well-characterized antibodies: glial fibrillary acidic protein (GFAP) rabbit polyclonal (Dako, Glostrup, Denmark) 1:2000; CD68 monoclonal clone KP1 (Dako) 1:15,000; APP monoclonal clone 22C11 (Millipore, Billerica, MA) 1:300, and neurofilament-200 kDa (NF-200 kDa) monoclonal clone RMO-24 (Invitrogen) 1:1000 to label astrocytes, microglia, and axonal damage, respectively. Sections were preheated 30 min at 60°C and dewaxed by two successive 5-min baths in xylene before microwave treatment for antigen retrieval. Endogenous peroxidase activity was inhibited with a solution containing 10% methanol and 1% H2O2 for 5 min at room temperature, then the sections were blocked for 1 h at room temperature in the following solution: 5% normal serum, 1% bovine serum albumin, 0.1% Tween20 in phosphate-buffered saline. The primary antibody was incubated overnight at 4°C, and then sections were incubated with the appropriate secondary biotinylated-antibody (1:500) for 1 h at room temperature. This was followed by the addition of the ABC kit reagents (Vector Laboratories, Burlingame, CA) for 30 min at room temperature before being visualized using DAB (Vector Laboratories).
Cortical density analysis
Semiquantitative analysis of the immunohistochemistry experiments was performed by three different individuals blinded to the patients' medical history. Labeled cells on frontal cortex sections (two serial sections per case) immunostained with GFAP and CD68 antibodies were counted to determine whether the number of reactive astrocytes and microglia was different between the contused and the contralateral noncontused regions. Counting was completed over three fields at 20 × magnification for each brain section, totaling an area of 1.5 mm2 and using the AnalySIS LS Research software (Olympus Soft Imaging Solutions GmbH, Münster, Germany).
Axonal damage assessment
Axonal damage was detected using immunohistochemistry for APP and NF-200 kDa proteins on sections including the corpus callosum as described above. Sections were analyzed without knowledge of the survival time or medical history. Typical features of axonal damage included the presence of axonal bulbs, swelling, and varicosities. Diffuse injury was defined by the presence or lack of immunostained damaged axons by both antibodies. There was no immunoreactivity in normal axons using this method.
Statistics
Statistical analysis was performed using SigmaStat software (SPSS Inc., Chicago, IL). Kolmogorov–Smirnov test (with Lilliefors' correction) was used to test data for normality within each group and values were transformed by natural logarithm calculation if required. One-way ANOVA was followed by multiple comparisons using the Student–Newman–Keuls method or Student's t test to identify significant difference between trauma and control groups.
Results
In situ pro-inflammatory cytokine protein levels are increased after TBI
To better understand the inflammatory cascade following injury in the human brain, we measured the protein levels of several inflammatory cytokines in post mortem cortical tissue homogenates. Six pro-inflammatory cytokines (IL-6, IL-1β, TNF-α, IFN-γ, GM-CSF, and IL-2), two cytokines with anti-inflammatory properties (IL-4 and IL-10) and one chemokine (IL-8) were analyzed. All the pro-inflammatory mediators measured were found to be markedly and significantly increased (p < 0.001) in the brain samples from the late group compared with control brains as well as the levels detected in the early group (Fig. 1). Among all cytokines, IL-6 showed the strongest increase, being of negligible levels in the control group (0.7 pg/mg of total protein), increasing to 1.4 pg/mg of total protein in the early group, and reaching the maximal concentration of 25.6 pg/mg of total protein in the late group (Fig. 1A). The second factor showing the strongest increase was the chemokine IL-8 with levels reaching 14.3 pg/mg of total protein in the late group (Fig. 1B), followed by IFN-γ (Fig. 1C) and TNF-α (Fig. 1D) with levels of 5.9 and 4.7 pg/mg of total protein, respectively. IL-1β (Fig. 1E) and GM-CSF (Fig. 1F) showed similar concentrations in the late group, being 2.4 and 2.2 pg/mg of total protein, respectively. The increase was very modest for IL-2 (Fig. 1G) but still statistically significant (0.2 pg/mg of total protein) as compared to undetectable amount in the control and early groups. Interestingly, in the samples from the early group lower cytokine concentrations were detected. However, IL-6 (p < 0.027), IFN- γ (p < 0.018), TNF-α (p < 0.003), and GM-CSF (p < 0.022) maintained a significant elevation over the non-TBI control group (Fig. 1A,C,D,F).
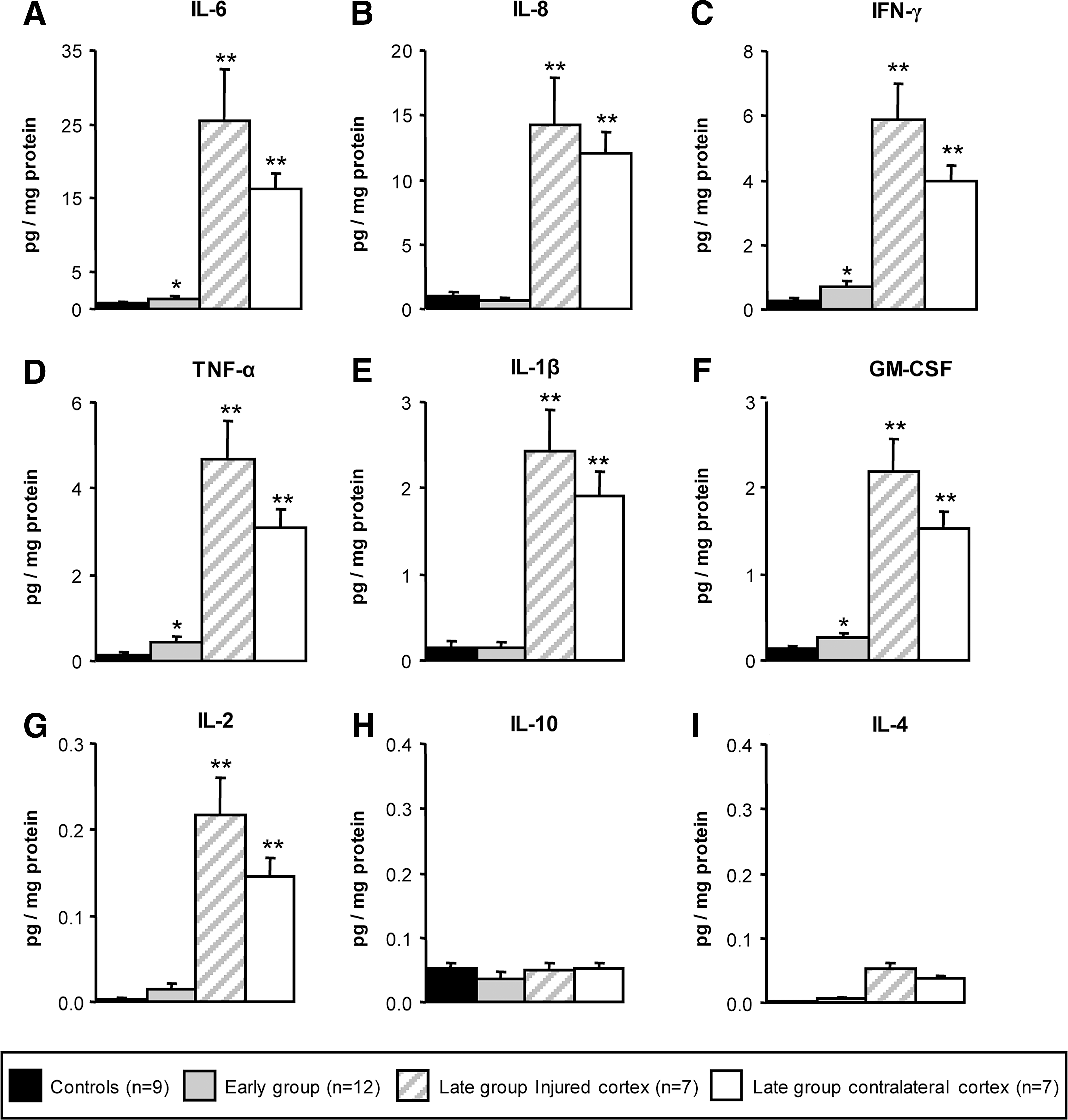
Inflammatory cytokine protein levels in the human cortex following TBI. All the pro-inflammatory mediators analyzed (IL-6, IL-8, IFN-γ, TNF-α, IL-1, GM-CSF, IL-2) (
In regard to the two anti-inflammatory cytokines IL-10 (Fig. 1H) and IL-4 (Fig. 1I), only concentrations below 0.05 pg/mL of total protein could be detected in the late group; however, particularly for IL-4, this modest increase was significantly higher when compared to the control group.
Interestingly, in the longer survival time brains, cytokine concentrations were elevated to a similar extent in the ipsi- and contralateral cortex, with no statistical difference between the two brain regions. The cytokine levels from the contralateral brain hemisphere were as high as the levels from the region in the vicinity of the macroscopic damage and statistically greater than the early groups (Fig. 1).
A linear regression analysis between the post mortem delays and the cytokine levels was performed to confirm there was no confounding relationship between the two (correlation of determination R 2 = 0.0244 and adjusted R 2 = 0.000).
Cytokine mRNA levels are also increased in human brain tissue
In order to establish whether the increase in cytokine protein levels corresponded to up-regulated mRNA expression in the traumatized human brain, we performed quantitative reverse-transcription PCR specific to IL-6, IL-1β, IL-8, and TNF-α. These four pro-inflammatory mediators showed a significant increase of their mRNA levels in the late group compared with the control group (Fig. 2).
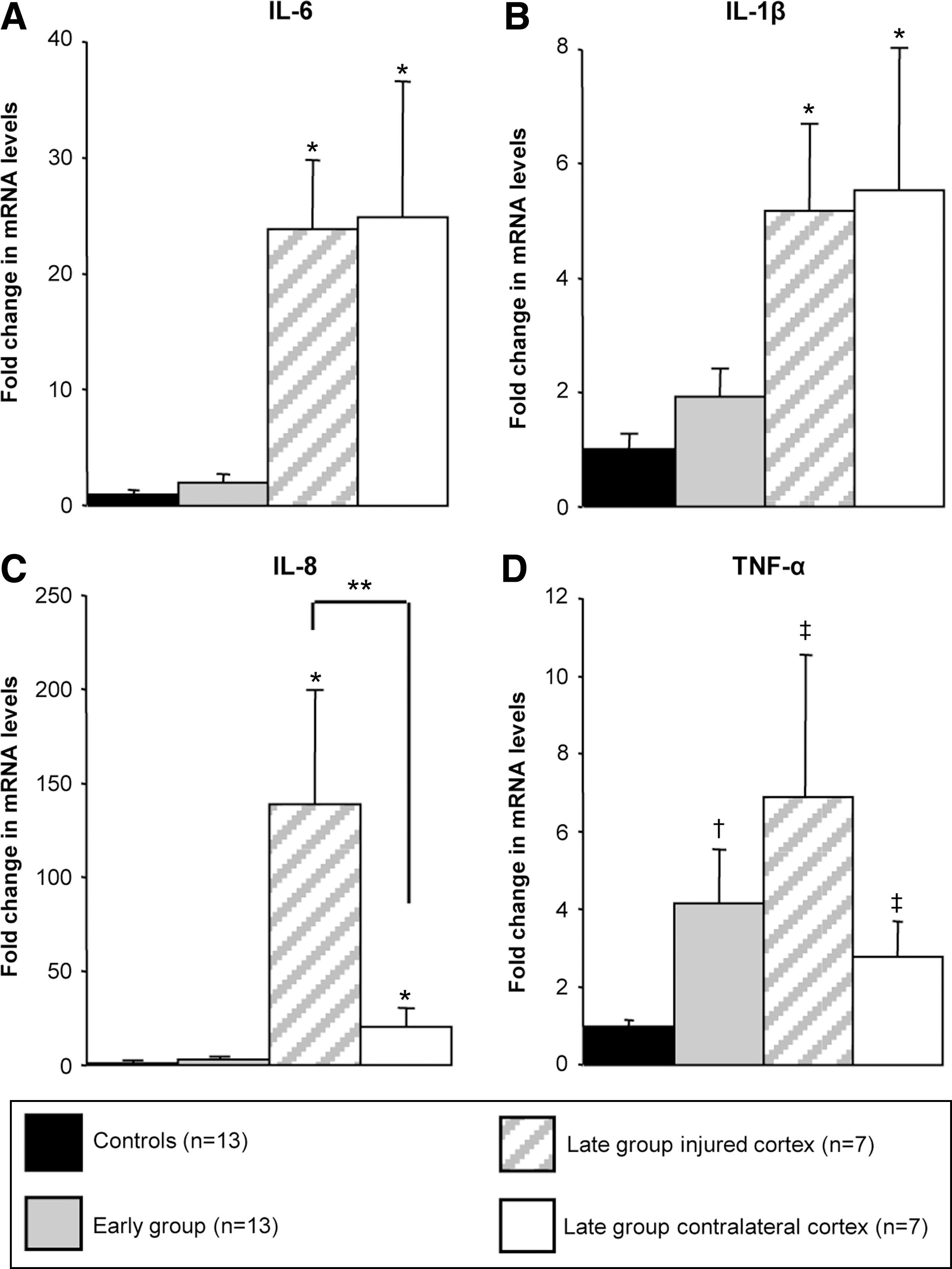
Cytokine mRNA levels in the human cortex following TBI. In the samples of individuals who died within 17 min of brain injury (gray bars) there was no difference in the levels of IL-6 (
IL-6 and IL-1β mRNA levels in the late group showed a 24-fold (p < 0.001) and 5-fold (p = 0.003) increase respectively, when compared to the control group, while no difference was detected between the ipsi- and contralateral regions. The mRNA level from the contralateral brain hemisphere was elevated at similar extent to the region in the vicinity of the injury (25- vs. 24-fold for IL-6, and 5.5- vs. 5.2-fold for IL-1β, respectively). Altogether, the IL-6 and IL-1β mRNA expression in the late group was significantly increased in comparison to the control and the early groups (Fig. 2A and 2B).
IL-8 is the chemokine showing the most substantial mRNA up-regulation in the late group with a 56-fold (p < 0.001) and a 139-fold (p < 0.001) increase in comparison to the early and control groups, respectively. Interestingly, IL-8 is the only mediator showing a statistical difference between the two brain regions analyzed. The mRNA levels were sevenfold lower in the contralateral brain hemisphere when compared to the mRNA levels from the region in the vicinity of the injury (p < 0.029). However, IL-8 mRNA levels were still 20-fold higher in the contralateral brain hemisphere when compared to the control (p < 0.001) or early groups (p = 0.001) (Fig. 2C).
TNF-α is the only cytokine tested showing a significant fourfold increase of its mRNA levels in the early group when compared to the control group (p = 0.014). The late group showed a further significant increase of mRNA expression when compared to the control group in the region close to the tissue damage with a sevenfold increase (p = 0.006) and in the contralateral brain hemisphere with a threefold increase (p = 0.035) (Fig. 2D).
Reactive astrocytes and microglia are present throughout the injured brain
Immunohistochemistry using antibodies against GFAP and CD68 was used to detect reactive astrocytes and microglia in the brain cortex following injury (Fig. 3). Semiquantitative analysis was performed to monitor cellular inflammation and compare the cortical region adjacent to the injury with the contralateral brain hemisphere in the late group. The number of positive cells counted for each case by the three different investigators was averaged and the values obtained were all within 10% of each other indicating low variability. Few GFAP- or CD68-positive cells were detected in the cortices of normal individuals and patients who died within 17 min of the injury (Fig. 3A,D). In marked contrast, a large number of reactive astrocytes and microglia was detected in the cortices of individuals who died more than 6 h after injury (Fig. 3B,E). However, no significant difference was detected in the number of positive cells between the two brain regions analyzed (GFAP: 95.93 ± 8.8 vs. 72.44 ± 15.19, p = 0.211 and CD68: 88.48 ± 27.43 vs. 64.74 ± 9.23, p = 0.431), indicating the presence of cellular inflammation throughout the injured brain within only a few hours following injury (Fig. 3C,F).
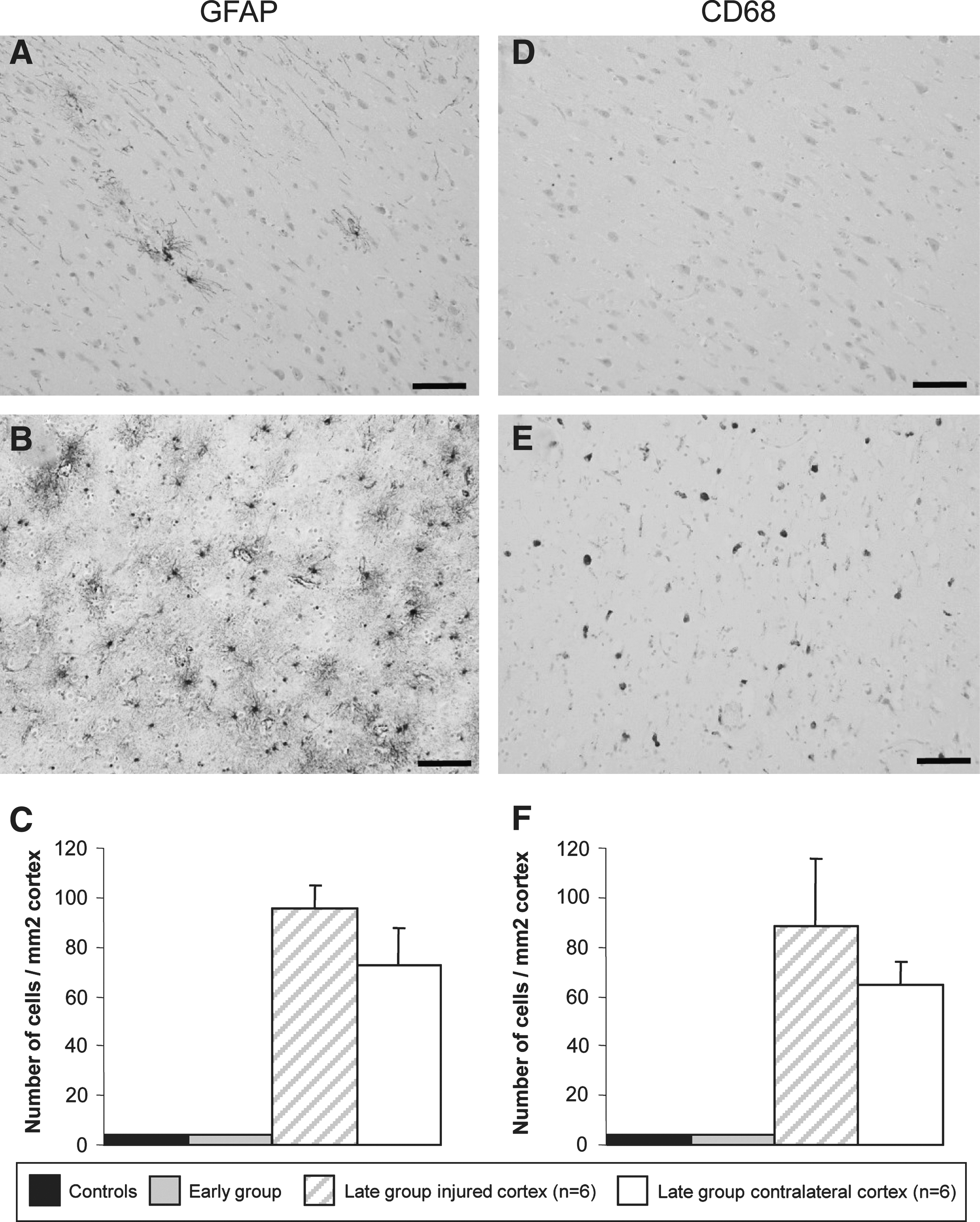
Density of activated astrocytes and microglia in the human cortex following TBI. GFAP and CD68 immunostaining of frontal cortex sections of a control individual (
Prominent axonal damage in all cases with late survival time
Axonal pathology of the 21 trauma cases was explored using immunohistochemistry against APP and NF-200 kDa proteins (Fig. 4) on brain sections containing the corpus callosum. The accumulation of these two proteins was observed as early as 8 h following injury (Fig. 4) and became more abundant over time (Fig. 4 and data not shown). We observed the typical features of axonal damage including axonal bulbs (Fig. 4C), swelling (Fig. 4F), and varicosities (Fig. 4B). Damaged axons were scattered within the white matter but were predominantly orientated along the axonal bundles (Fig. 4A,D). In addition, immunoreactive axons for APP showed similar patterns of positive NF stained axons (Fig. 4A,D). The consistent presence of numerous damaged axons indicated that diffuse brain injury was present in all the cases with a survival time of 8 h or longer. As expected, no staining was detectable for APP and NF in the brain samples of patients who died within 17 min of the injury or in the control individuals (data not shown).
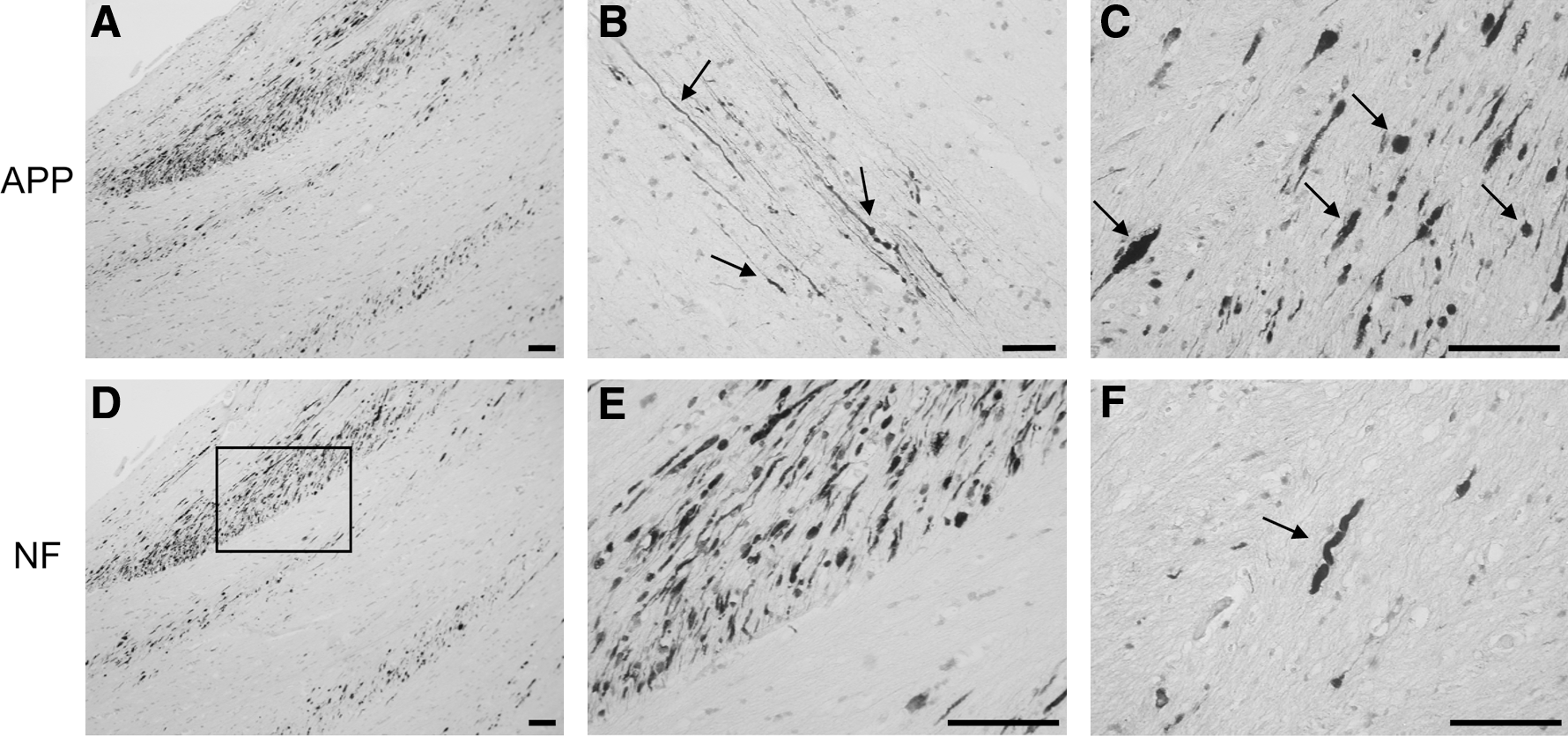
Axonal damage assessment in the corpus callosum of TBI patients. Amyloid precursor protein (APP) (
Discussion
The present study is the largest and first of its kind assessing humoral and cellular inflammation elicited after TBI in post mortem human brain tissue. The results presented in this manuscript confirm and extend previous observations on the inflammatory cascade following TBI in animal and human studies. Firstly, we demonstrated that pro-inflammatory cytokine protein levels of IL-1β, IL-2, IL-6, IL-8, IFN-γ, TNF-α, and GM-CSF were significantly increased within the brain tissue, following acute brain injury in humans. In marked contrast, the levels of anti-inflammatory cytokines IL-4 and IL-10 remain unchanged. Secondly, the cerebral inflammatory response was triggered within a few minutes of acute TBI as shown by the significant increase of IL-6, IFN-γ, TNF-α, and GM-CSF protein levels in the early group. Thirdly, this is the first quantitative mRNA study on human brain tissue in which the mRNA levels of several inflammatory mediators, including IL-6, IL-8, TNF-α, and IL-1β, were shown to be significantly increased following TBI. Fourthly, the raised cytokine protein levels coincided with the increased levels of their mRNAs, indicative of an immediate cerebral inflammatory response. Finally, evidence of diffuse axonal injury corresponded with a robust cytokine inflammatory response, independent of the macroscopic injury pattern.
More than 20 years ago Young's group firstly showed an increase in the activity of IL-1 (McClain et al., 1987) and production of IL-6 (McClain et al., 1991) in the CSF of TBI patients the day of their hospital admission. Following this work, our group later characterized longitudinal patterns of cytokine production (TNF, IL-6, IL-8, IL-10, TGF-β) in patients with severe TBI up to 3 weeks post-injury (Csuka et al., 1999; Kossmann et al., 1995, 1996, 1997; Morganti-Kossmann et al., 1999; Pleines et al., 2001). The most interesting finding of all these studies was that the cytokine levels detected in the CSF were far higher than those detected in blood serum, suggesting that neural cells themselves were the source of intrathecal cytokine production. These early findings also demonstrated that the secretion of cytokines in the CSF remained elevated for a longer period of time as compared with rodent brain. The data on human brain tissue presented in this study confirm unequivocally this long-standing assumption.
IL-6 has been one the most investigated cytokines since it is the main inducer of the acute phase reaction and because it was found to be released in the largest amounts, as compared to other cytokines (TNF-α, TGF-β, IL-10), in the CSF of TBI patients (Csuka et al., 1999). Consistently in this study, we also found that IL-6 is the cytokine produced at highest level of expression following injury in the human brain tissue. In TBI patients from the late group (mean survival time of 40 h), IL-6 mRNA and protein levels were found to be up to 25-fold higher than control and early groups (Figs. 1A, 2A). Interestingly, in the early group, IL-6 protein levels had already doubled, indicating an immediate response to the injury, thus confirming its neural production in the human brain.
IL-1β is another cytokine frequently studied because of its role as a major promoter of neuroinflammation. Several studies have reported elevation of IL-1 at protein and mRNA levels in animal models of TBI (Fan et al., 1995; Fassbender et al., 2000; Folkersma et al., 2008; Kamm et al., 2006; Woodroofe et al., 1991) and in the CSF or microdialysates of TBI patients (Buttram et al., 2007; Holmin and Höjeberg, 2004; Hutchinson et al., 2007; Mellergard et al., 2008; Winter et al., 2002). A few studies in human brain tissue samples obtained from TBI patients who underwent a decompressive craniectomy have detected expression of IL-1β following brain injury. In particular, a study by Clark and colleagues showed that caspase-1 (or IL-1 converting enzyme) is also activated post-injury (Clark et al., 1999). This enzyme cleaves pro-IL-1 to generate active IL-1β. Also, a study by Holmin and Höjeberg (2004) demonstrated a strong expression of IL-1β within pericontusional tissue collected intra-operatively 24 h after injury, indicating a rapid inflammatory response. As the region analyzed is very small and close to the injury site, these studies may not reflect what occurs in the injured brain as a whole. Altogether they were the first to describe a temporal regulation of IL-1β in the human brain tissue. Here we have complemented the previous studies by demonstrating that IL-1β mRNA and protein levels are on average five times more elevated in brain tissue of TBI patients than in the control samples and that the raised IL-1 mRNA is detected in the early hours and maintained for several days after TBI (Figs. 1E, 2B). Our data therefore confirm and reinforce the results from previous studies showing a rapid expression and activation of IL-1β following brain injury.
TNF-α is a small 17-kDa peptide expressed by astrocytes, microglia, and neurons. Its action is mediated via binding to TNF receptors located on glia and neurons (Merrill, 1991; Wolvers et al., 1993). TNF-α, like IL-6 and IL-1β, is a key initiator of the inflammatory response and has been extensively analyzed mainly in animal models of TBI (Fan et al., 1996; Knoblach et al., 1999; Shohami et al., 1994; Taupin et al., 1993) but also in the CSF and serum of TBI patients by our group (Csuka et al., 1999) and others (Hayakata et al., 2004; Shiozaki et al., 2005). Until now, no in situ data on the expression of TNF-α in human brain following TBI was available. In this study we report that TNF-α is the only inflammatory mediator showing a significant increase at both mRNA (fourfold) and protein (threefold) levels in the early group (Figs. 1D, 2D). This finding correlates with the increased expression of caspase-8 (linked with TNF receptor activation), predominantly observed in neurons of human brain tissue samples obtained from TBI patients who underwent a decompressive craniectomy (Zhang et al., 2003). Altogether these data demonstrate that different players of the early inflammatory response are activated in the injured brain immediately and locally in response to trauma.
Chemokines play a critical role in the neuroinflammatory response by initiating the recruitment of peripheral leukocytes following TBI. IL-8, also known as CXCL8, is a powerful chemotactic factor for neutrophils and was shown to be elevated in the CSF of both adults and children following TBI (Kossmann et al., 1997; Whalen et al., 2000). A recent study by Stefini and colleagues showed that IL-8 is also highly expressed in the tissue of patients with brain contusions (Stefini et al., 2008). Our results showed a 139-fold increase in IL-8 mRNA levels in brain cortex taken in the vicinity of the injury while a lower 20-fold increase was observed in the contralateral brain hemisphere concordant to the raised protein level. Interestingly, no difference between those two regions was detected in regards to IL-8 protein level, both showing a significant increase when compared to either the control brain or early TBI groups (Figs. 1B, 2C). One possible explanation is that in post mortem brain tissue protease activity is more elevated in the proximity of the injury, consequently altering the cytokine levels, in contrast to the contralateral cortex where cytokine measurements were very similar. However, this was different for IL-8 compared to other cytokines. What our results also suggest is that IL-8 expression may be more specifically up-regulated in the proximity of the local injury and this hypothesis is supported by animal studies showing abundant accumulation of neutrophils already at 1 day post-injury (Stahel et al., 2000; Clark et al., 1994).
The pro-inflammatory mediators IL-1β, IL-2, IL-6, IL-8, GM-CSF, IFN-γ, and TNF-α have very similar patterns of expression post-injury with higher protein levels mostly detected in the late group. Interestingly, IL-6, TNF-α, GM-CSF, and IFN-γ protein concentrations are already significantly increased a few minutes post-injury, indicating their important role in the early phases of the inflammatory cascade. This result also suggests that these cytokines are locally expressed by neuronal and/or glial cells and their paracrine mode of action may be important to propagate the inflammatory response. This work highlights the need to investigate further the cellular expression of these factors to identify their source of production following injury. In marked contrast, the concentrations of the anti-inflammatory cytokines IL-4 and IL-10 remained at basal levels in both TBI groups. This is in conflict to early production of IL-10 in CSF of adult and pediatric TBI (Bell et al., 1997; Csuka et al., 1999).
Another interesting and surprising observation was the absent differences in the cytokine protein and mRNA levels between the two brain regions analyzed: the first within the cortex, proximal to the injury site, and the second in the contralateral cortex in a region with no morphological signs of tissue damage. GFAP and CD68 immunohistochemistry showed an extensive distribution of reactive astrocytes and microglia, with no difference between these two regions (Fig. 3). In addition, APP and NF immunoreactivity illustrated diffuse axonal injury in each case within the late group indicating a profound axonal damage (Fig. 4). Altogether these results suggest that the extensive inflammation is not only occurring at the focal injury site, which was detected macroscopically during the pathological analysis, but is also present within the diffuse axonal injury regions of the brains analyzed in our study.
While every effort was made to generate homogeneous groups, it is important to note when studying post mortem human brain tissue in neurotrauma that the location of the sample within the brain is a limiting factor. TBI patients are a very heterogeneous group by nature due to the different proportions of focal and diffuse brain damage. Furthermore, the influence of secondary factors like respiratory distress, hypoxia, or drop in systemic blood pressure (hypotension), often observed in severe multitrauma patients, can generate secondary brain damage such as ischemic injuries that exacerbate both morphological and biochemical changes. In addition, the blood–brain barrier disruption may also influence the protein levels detected in the brain tissue including the inflammatory mediators triggered in severe multitrauma patients. Nevertheless, the present study extends previous observations on the release of cytokines in the CSF of severe TBI patients by clearly showing for the first time in human brain tissue that 1) the inflammatory response begins immediately after the traumatic impact; 2) diffuse secondary axonal injury may contribute to the extent of cellular and humoral neuroinflammation throughout the entire brain; and 3) cytokines and chemokines detected in the brain tissue are produced locally by intraparenchymal cells of the central nervous system in the early stages of the inflammatory cascade and do not diffuse from the systemic circulation. Characterization of the cellular source of these factors is therefore needed in order to understand the mode of action of each of the cytokines following severe brain injury in humans.
Footnotes
Acknowledgments
We thank Dr. Edwin Yan for his help with the Bio-plex cytokine assays and Dr. Nicole Bye for her valuable comments on the manuscript. This study was supported by the Victorian Neurotrauma Initiative. Tissues were received from the Victorian Brain Bank Network, supported by The National Trauma Research Institute, The University of Melbourne, The Mental Health Research Institute of Victoria, The Victorian Institute of Forensic Medicine and funded by the Victorian Neurotrauma Initiative, Neurosciences Australia, and the National Health and Medical Research Council of Australia.
Author Disclosure Statement
No conflicting financial interests exist.