
Abstract
Traumatic brain injury (TBI) is one of the most acute degenerative pathologies in the central nervous system, and in vivo indices enabling an assessment of TBI on a mechanistic basis have yet to be established. The aim of this work was to pursue neuroinflammatory changes and their link to functional disruptions of traumatically-damaged neurons in a rat model of TBI by longitudinal positron emission tomographic (PET) assays. TBI was induced in the unilateral frontal cortex of craniotomied rats according to a lateral fluid percussion brain injury protocol. The use of [18F]fluoroethyl-DAA1106 as a PET tracer for translocator protein (TSPO) permitted demonstration of the inflammatory response to the injury, peaking at 1 week after impact. This alteration was parallel to metabolic deficits assessed by PET with [18F]fluorodeoxyglucose, but the difference in TSPO levels between impacted and non-impacted frontal cortices was more than threefold of the interlateral metabolic difference, indicating superiority of TSPO imaging for sensitive detection of post-traumatic pathologies. Comparative PET, autoradiographic. and immunohistochemical investigations illustrated the primary contribution of hypertrophic microglia and macrophages to acute TSPO signals in the vicinity of the impact. Astrocytes also formed a TSPO-positive glial scar encompassing necrotic inflammation, and were clustered with PET-detectable TSPO signals in the bilateral external and internal capsules at late stages, putatively reacting with diffuse axonal injury. These observations support the applicability of TSPO-PET as an imaging-based preclinical and clinical biomarker assay in TBI, and indicate its potential capability to clarify aggressive and protective roles of glial responses to injury when combined with emerging anti-inflammatory and immunomodulatory treatments.
Introduction
N
Neuroinflammatory responses to TBI have drawn biomedical interest in the form of diagnostic and therapeutic targets (Laird et al., 2008; Lloyd et al., 2008; Morganti-Kossmann et al., 2007), and in vivo visualization of immunocompetent cells activated in the brain by pathological stimuli has been enabled by PET radioligands for 18-kDa translocator protein (TSPO). This molecular component becomes overexpressed at the outer mitochondrial membrane of monocytes, macrophages, microglia, and astrocytes concurrently with their participation in diverse neuropathologies (Chen et al., 2004; Myers et al., 1991; Raghavendra Rao et al., 2000; Stephenson et al., 1995). PET assessments of neuroinflammatory changes in patients with neurodegenerative diseases (Cagnin et al., 2001; Ouchi et al., 2005), and experimental models of these disorders (Cicchetti et al., 2002; Rojas et al., 2007; Venneti et al., 2009), were initially established with a 11C-labeled TSPO imaging agent, [11C]PK 11195. Subsequently, our animal and human PET studies have demonstrated that new classes of TSPO-binding compounds, including the DAA1106 family (Maeda et al., 2004; Yasuno et al., 2008; Zhang et al., 2004), possess reasonably high blood–brain barrier (BBB) permeabilities and affinities for TSPO. We have also revealed the contribution of both microglia and astrocytes to in vivo signals of radiolabeled DAA1106, and the functional implications of these TSPO-positive glial cells in neurotoxic injuries (Ji et al., 2008; Maeda et al., 2007a). Moreover, good performance of [11C]DAA1106 in detecting inflammation in a rat model of TBI has been shown in a recent PET experiment (Venneti et al., 2007).
The present rat PET study aimed to assess the significance of TSPO imaging in clarifying the roles of neuroinflammatory cells in the disruption and restoration of neuronal integrity following mechanistic injuries. TSPO changes indicating neuronal viability and recoverability were pursued by the combined use of [18F]fluoroethyl-DAA1106 ([18F]FE-DAA11060) and [18F]FDG in the same individual animals. Cellular localization of TSPO signals was also immunohistochemically investigated in order to gain insights into deleterious versus beneficial functions of glial cells, as demonstrated in our previous study of nontraumatic conditions (Ji et al., 2008).
Methods
Animals
Adult male Wistar rats initially weighing 230–250 g were used. Three or four rats were kept together in each cage. All animals studied here were maintained and handled in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals and our institutional guidelines. Protocols for the present animal experiments were approved by the Animal Ethics Committees of the National Institute of Radiological Sciences.
Lateral fluid percussion brain injury
We induced TBI in rat brains by means of a lateral fluid percussion brain injury (FPBI) protocol, which is an extensively characterized and widely used preclinical model of human closed-head injury (Thompson et al., 2005). The animals were anesthetized with a single intraperitoneal injection of sodium pentobarbital (60 mg/kg), and were placed in a stereotaxic frame (Narishige, Tokyo, Japan). A 3-mm-diameter craniotomy was made 2.5 mm posterior to the bregma and 2.5 mm lateral to the midline on the right convexity. TBI was produced by a fluid percussion device (model HPD-1700; Dragonfly, Ridgeley, WV). A brief (20- to 25-msec), transient pressure fluid pulse impact was applied to the exposed dura. Pressure pulses were measured extracranially by a transducer and recorded on a storage oscilloscope. The force of the impact was adjusted to 1.61 ± 0.14 atm. Analgesics were not used for these animals in the peri- and postoperative stages. A total of 21 rats in two cohorts received fluid percussion injury. The first group consisted of eight animals, and two died of cardiorespiratory arrest immediately after the impact. Three recovered from post-traumatic respiratory arrest over several seconds, and three displayed no noticeable complications. All six survivors were then used for longitudinal TSPO-PET analyses, but one of them was eliminated from the study group due to a failure of the radiotracer injection in the first scan. The second group included 13 rats, three of which died of cardiorespiratory arrest immediately after the impact. Respiratory arrest also occurred in four other individuals, and they recovered in several seconds. Six animals did not exhibit any perioperative complications. All 10 survivors were subsequently employed for PET imaging. Six rats including two animals with transient respiratory arrest were longitudinally assessed by TSPO and metabolic PET scans, and four other rats including two animals with transient respiratory arrest were employed for verapamil-PET at 1 week after the impact. None of the survivors of the surgery exhibited additional complications that impeded the assays.
We also used six untreated rats without any operative procedures as controls. The validity of employing these naïve rats was examined in our pilot study by comparing their [18F]FE-DAA1106-PET data with those of rats at 1 week after sham operation in a smaller cohort (n = 3), and the radiotracer uptake in these two groups was found to be nearly identical.
Radiotracer preparation
[18F]FE-DAA1106 was radiosynthesized using its desmethyl precursor, DAA1123, which was generously provided by Taisho Pharmaceutical (Tokyo, Japan), as described in detail elsewhere (Zhang et al., 2004; Maeda et al., 2007b). The radiochemical purity of the end product exceeded 95%, and the specific radioactivity was 120 ± 20.5 Gbq/μmol (mean ± SD) at the end of synthesis. [18F]FDG was purchased from Nihon Medi-Physics (Tokyo, Japan), and the specific radioactivity at the time of injection was 50–70 Gbq/μmol.
[11C]verapamil was synthesized from norverapamil (Eisai, Tokyo, Japan) as described previously (Wegman et al., 2002), and was diluted with 2–3 mL of saline containing 0.75% polyoxyethylenemonosorbitan oleate and 1% ascorbic acid. The radiochemical purity was more than 95%, and the specific radioactivity ranged from 28.3–79.7 Gbq/μmol (mean ± SD: 47.6 ± 17.3 Gbq/μmol).
In vivo small-animal PET scans
Neuroanatomical template images of the rat brain consisting of axial T1-weighted MR images were generated by a 7-tesla high-resolution magnetic resonance imaging (MRI) system (NIRS/KOBELCO, Kobe, Japan/Bruker BioSpin GmbH, Ettlingen, Germany). PET assessments of neuroinflammation, glucose metabolism, and BBB integrity were conducted for rats given TBI. Eleven rats underwent PET assays of TSPO using [18F]FE-DAA1106 at 1, 4, and 9 weeks after injury. Six of these animals were also studied using [18F]FDG along with each [18F]FE-DAA1106-PET measurement. Additionally, four animals scanned with these two tracers were assayed with [11C]verapamil at 1 week. Since [11C]verapamil is a good substrate for an efflux transporter, p-glycoprotein, in cerebrovascular endothelial cells, it is poorly permeable through undamaged BBB, enabling evaluation of the BBB intactness in living brains (Ikoma et al., 2006). [18F]FDG-PET analyses were also applied to 11 uninjured rats as controls, and six of these animals subsequently underwent [18F]FE-DAA1106-PET scans. The examinations with different tracers were carried out 1–2 days apart. All scans were performed using a microPET Focus 220 animal scanner (Siemens Medical Solutions USA, Knoxville, TN) designed for rodents and small monkeys, which provides 95 transaxial slices 0.815 mm (center-to-center) apart, a 19.0-cm transaxial field of view (FOV), and a 7.6-cm axial FOV (Tai et al., 2005). Prior to the PET scans, the animals were anesthetized with 1.5% isoflurane. Data acquisition in a 3D list mode with an energy window of 350–750 keV began 40 min after the intravenous injection of [18F]FE-DAA1106 via the tail vein as a single bolus (imaging time 20 min; mean dose ± SD, 55.6 ± 19.2 Mbq). Similarly, [18F]FDG-PET scans were initiated at 60 min after radiotracer injection (imaging time, 10 min; mean dose ± SD, 71.4 ± 2.7 Mbq), and imaging was started immediately after the administration of [11C]verapamil (imaging time, 10 min; mean dose ± SD, 103.2 ± 5.9 Mbq). All list-mode data were sorted and Fourier-rebinned into 2D sinograms. Images were thereafter reconstructed using 2D filtered back-projection with a 0.5-mm Hanning filter. Volumes of interest (VOIs) were placed on multiple brain areas using PMOD® image analysis software (PMOD Group, Zurich, Switzerland) with reference to the MRI template (Fig. 1). Tracer uptake in each VOI was estimated as the percentage of injected dose per tissue volume (%ID/mL), and standardized uptake value (SUV), which was defined as: radioactivity (Mbq/mL)/[injected dose (Mbq)/body weight (g)].

Coronal [18F]fluoroethyl-DAA1106 PET images in a rat at 1 week after fluid percussion injury, illustrating the definition of volumes of interest (VOIs) in the frontal cortex (FC) and striatum (ST). Distances from the bregma are indicated at the bottom of the images (PET, positron emission tomography).
In vitro autoradiographic analysis of neuroinflammatory changes
The rats were deeply anesthetized with sodium pentobarbital, and were transcardially perfused with phosphate-buffered saline at different time points after TBI. Brain tissues were then removed and fixed with 4% paraformaldehyde in phosphate buffer. Then 10-μm-thick frozen brain sections were created with a cryostat (HM560; Carl Zeiss, Jena, Germany), and coronal sections of rat brains taken 2.5 mm posterior to the bregma were picked up as representative samples, so the data could include the hippocampus, striatum, thalamus, internal and external capsules, and corpus callosum as well as the necrotic core around the impact site. [18F]FE-DAA1106 was synthesized as in the PET measurements. Autoradiographic assays of TSPO were conducted using paraformaldehyde-fixed frozen sections. The transcardial perfusion and subsequent fixation did not noticeably affect the regional profile and intensity of in vitro [18F]FE-DAA1106 signals (data not shown), as in our previous study (Ji et al., 2008). The samples were pre-incubated in 50 mM Tris-HCl buffer (pH 7.4) for 15 min at room temperature, followed by reaction with [18F]FE-DAA1106 (18.5 Mbq/L; 0.35 nM) in 50 mM Tris-HCl buffer (pH 7.4) for 60 min at 25°C. Nonspecific binding of the radioligands was determined in the presence of 10 μM PK11195 (Sigma-Aldrich, St. Louis, MO) in the same reaction solution. After incubation, they were rinsed with ice-cold water for 10 sec, warmly blow-dried and placed on an imaging plate (BAS-MS2025; Fuji Film, Tokyo, Japan) for 1 h. Radiolabeling was detected by scanning the imaging plate using the BAS5000 system (Fuji Film).
Immunohistochemical analyses
The paraformaldehyde-fixed frozen brain sections subadjacent to the autoradiographic samples were immunostained based on a standard protocol using fluorophore-conjugated secondary antibodies. For TSPO staining, the sections were autoclaved beforehand at 121°C for 5 min in 10 mM sodium citrate buffer (pH 6.0) for antigen recovery. Nonspecific binding of antibodies was blocked by treating sections with blocking reagent supplied in a Tyramide Signal Amplification system (TSA Biotin System; PerkinElmer, Waltham, MA). Primary antibodies were diluted in Tris-buffered saline (TBS; 0.1 M Tris-HCl and 0.15 M NaCl [pH 7.5]), and were reacted with the sections overnight at 4°C. After being rinsed three times in TBS containing 0.05% Tween 20, the sections were incubated with Alexa Fluor 488- and Alexa Fluor 568-conjugated anti-IgG secondary antibodies (Invitrogen, Carlsbad, CA) in TBS containing the above-mentioned blocking agent at room temperature for 1 h. All stained sections were cover-slipped using Vectashield Fluorescent Mounting Media (Vector Laboratories, Burlingame, CA), and were examined using an all-in-one microscope/digital camera (BZ-9000; Keyence, Osaka, Japan). The antibodies used in this study were as follows: rabbit polyclonal antibody against the C-terminal portion of murine TSPO (NP155; 1:500 dilution; Ji et al., 2008); rabbit polyclonal antibody against ionized calcium binding adapter molecule-1 (Iba-1; 1:500 dilution) (Wako Pure Chemicals, Osaka, Japan), recognizing microglia; and rabbit polyclonal antibody against glial fibrillary acidic protein (GFAP), recognizing astrocytes (1:500 dilution; Dako, Carpinteria, CA).
Statistical analysis
Effects of time points and regions on radiotracer uptake values (%ID / ml and SUV) and right-to-left ratio of radioactivity were assessed by Friedman test. These values in injured rats were also statistically compared with those in uninjured controls by Mann-Whitney U test with Bonferroni correction. Correlations between two parameters were examined by Spearman's rank correlation test.
Results
In vivo PET assessments of inflammatory and metabolic changes
Longitudinal PET scans with [18F]FE-DAA1106 and [18F]FDG illustrated the time course of neuroinflammatory activation parallel to metabolic disturbances in the brains of rats receiving mechanical impact (Fig. 2A). TSPO levels in the impacted frontal cortex were profoundly increased at 1 week, and thereafter appeared to descend during the next 8 weeks of observation. The difference in [18F]FE-DAA1106 binding between impacted and non-impacted cortices was clearly visible at 1 and 4 weeks, but was barely recognizable at 9 weeks. Intensification of TSPO signals was also observed in the striatum ipsilateral to the traumatic lesion at 1 and 4 weeks. Although reduced [18F]FDG uptake in the impacted frontal cortex was evident at 1 week, no overt metabolic alterations were detectable at 4 and 9 weeks, according to visual inspection of the images. A slight attenuation of [18F]FDG signals was also noted in the ipsilateral striatum. At 1 week after physical injury, enhanced accumulation of [18F]FE-DAA1106 spatially coincident with metabolic deficits was not accompanied by increased [11C]verapamil uptake (Fig. 2B). Indeed, averages of the maximal [11C]verapamil uptakes stayed at a very low level, and were 0.13% of the injected dose per unit volume (%ID/mL) in the right and left frontal cortices, and 0.1% ID/mL in the right and left striata (n = 4). There was also no right-left asymmetry in these [11C]verapamil measures. Thus we concluded that the rise of [18F]FE-DAA1106 radiosignals was not attributable to acceleration of radiotracer delivery induced by BBB disruptions.

PET images of rat brains given mechanical impacts on the right frontal cortex. (
[18F]FE-DAA1106 retention quantitatively estimated as %ID/mL was markedly elevated in the bilateral frontal cortices, with ipsilateral dominance at 1 week. Increases in radiosignals were statistically significant only at this time point compared with noninjured controls (Fig. 3A). The uptake values were then converted to SUVs based on body weights of the animals (mean ± SD: 245.6 ± 7.5, 281.5 ± 10.1, and 322.5 ± 9.8 g, at 1, 4, and 9 weeks, respectively). Unlike body mass–uncorrected uptake values, SUVs were significantly increased in both the ipsilateral and contralateral frontal cortices throughout the observation period, except for the contralateral side at 4 weeks (Fig. 3B). TSPO upregulation was also assessed by calculating right-left asymmetries of [18F]FE-DAA1106 distributions, and radioactivities were pronouncedly augmented in the impacted frontal cortex relative to the contralateral region at 1 and 4 weeks (Fig. 3C). Despite the lower magnitude of lateralities, the radiosignals in the striatum were consistently more intense on the injured side than in the opposite hemisphere during the time course of PET assays (Fig. 3C). Similar changes were also observed in six rats undergoing both TSPO and metabolic PET scans (Fig. 3D). Radioligand uptake determined as %ID/mL was significantly increased by 35% in the right and left thalamic ROIs at 1 week after impact compared with control measures (p < 0.05 by Mann-Whitney U test). Tracer accumulation in the brainstem at 1 week was also higher than control values by 25%, but this difference was not statistically significant (p > 0.05 by Mann-Whitney U test). Radiosignal intensities in these regions at 4 and 9 weeks were nearly identical to those seen in controls.
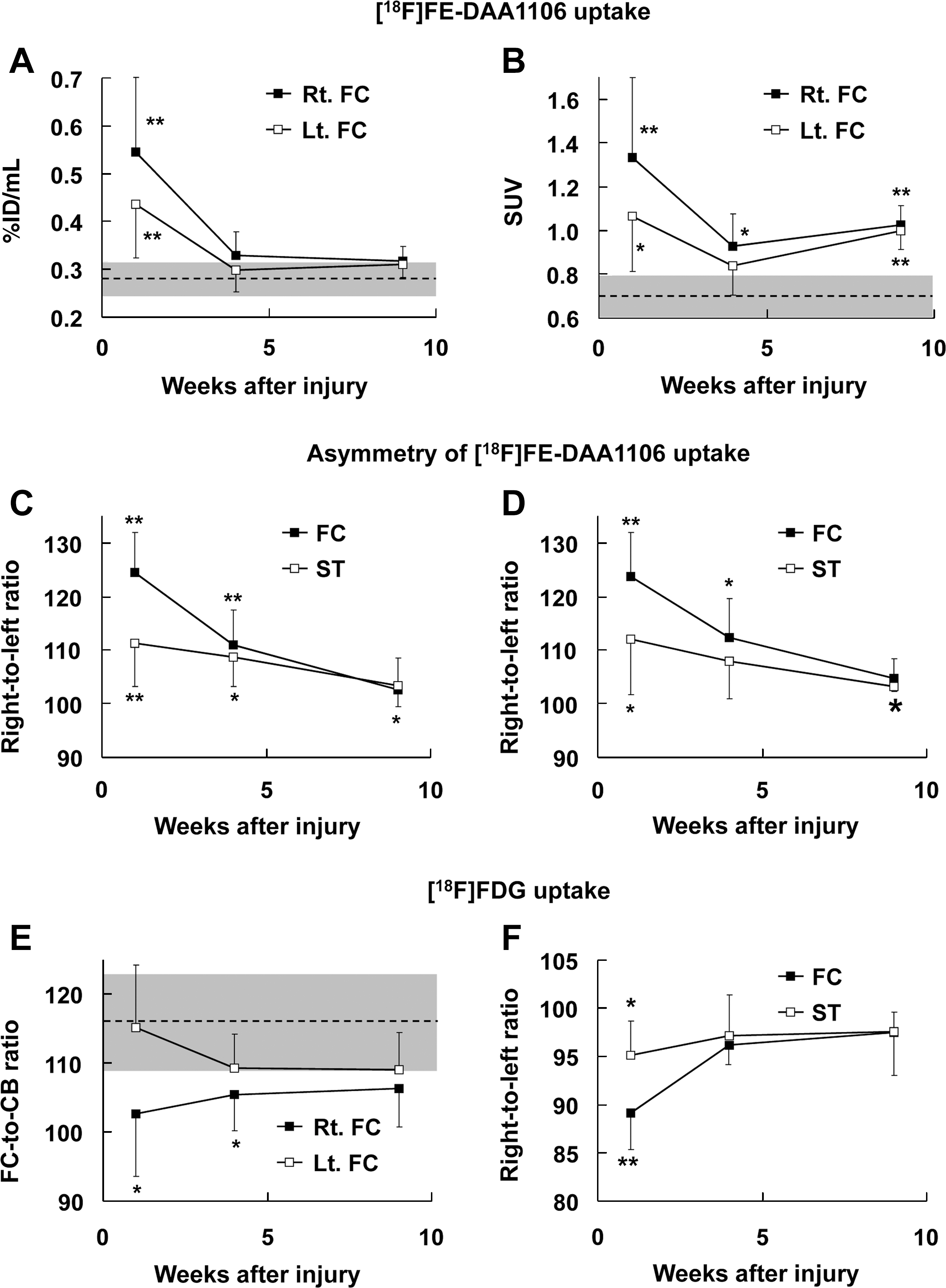
Quantitative assays of [18F]fluoroethyl-DAA1106 ([18F]FE-DAA1106) and [18F]fluorodeoxyglucose ([18F]FDG) uptake in rat brains at 1, 4, and 9 weeks after traumatic injury. (
In these animals, there was a large degree of interindividual variability of [18F]FDG uptake calculated as %ID/mL and SUV (data not shown), while metabolic ratios normalized by cerebellar radioactivities were significantly diminished in the ipsilateral frontal cortex at 1 and 4 weeks compared with nonimpacted control animals (Fig. 3E). Likewise, a remarkable reduction of glucose utilization in the impacted frontal cortex relative to its contralateral counterpart occurred only at 1 week (Fig. 3F). Metabolic asymmetry was also noticeable in the striatum at 1 week, although the changes in this area were less prominent than those in the frontal cortex (Fig. 3F). Alterations of [18F]FE-DAA1106 and [18F]FDG uptake at 1 week in the ipsilateral frontal cortex of these six animals were +24% and −11% of the contralateral measures, respectively, in the assessment of right-left asymmetries.
We further examined interactions between inflammatory and metabolic data in individuals assayed by both [18F]FE-DAA1106 and [18F]FDG. A significant correlation between ipsilateral-to-contralateral ratios of [18F]FE-DAA1106 and [18F]FDG uptakes was observed in the frontal cortex when all data at the three different time points were combined (Fig. 4A). According to the linear regression slope (−0.334), interlateral asymmetries of TSPO levels during the post-traumatic period were threefold greater than those of glucose utilizations. In addition to the overall correlation between inflammatory and metabolic indices, elevation of TSPO levels was associated with hypometabolism in the frontal cortex at 1 week (Fig. 4B). While the inflammatory laterality at 1 week was not linearly correlated with metabolic asymmetries at 9 weeks, it was remarkably profound in both of two animals showing an ipsilateral-to-contralateral metabolic ratio below control mean −2 SD at 9 weeks (Fig. 4B).
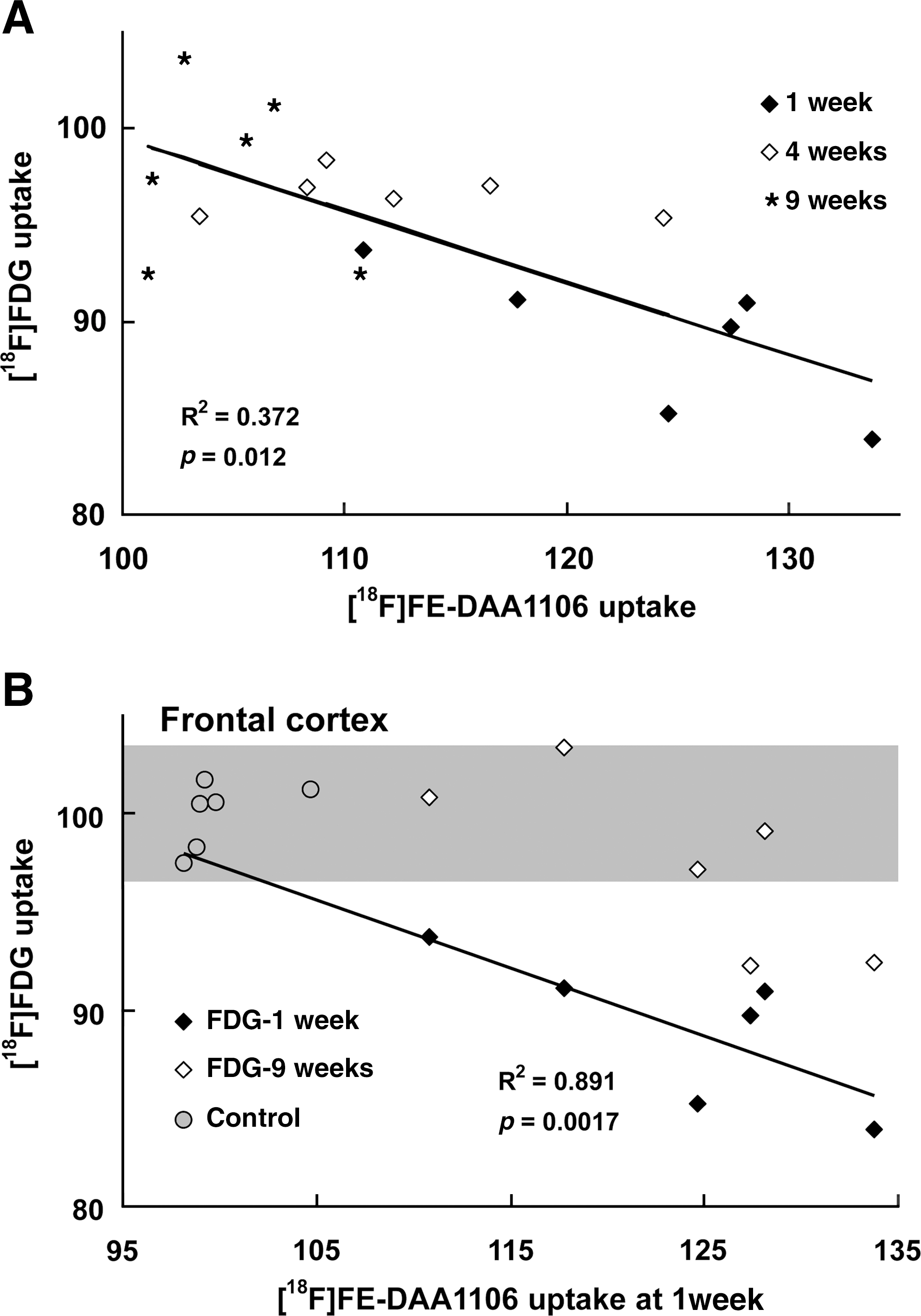
Interactions between [18F]fluoroethyl-DAA1106 ([18F]FE-DAA1106) and [18F]fluorodeoxyglucose ([18F]FDG) levels in injured rat brains. (
The time course of metabolic abnormalities was also dissected by examining the relationships between interlateral asymmetries of frontal [18F]FDG uptakes at different time points. From 1 to 4 weeks, ipsilateral-to-contralateral ratios of [18F]FDG signals displayed recovery to a uniform level in a manner unrelated to the severity of the initial metabolic deficits (R2 = 0.595 and p = 0.085 by Spearman's rank correlation test for the correlation between right-to-left ratios of [18F]FDG uptake in the frontal cortex at 1 and 4 weeks). Meanwhile, higher ratios at 4 weeks were significantly associated with more perturbed metabolic status at 9 weeks (R2 = 1.000 and p = 0.025 by Spearman's rank correlation test for the correlation between right-to-left ratios of [18F]FDG uptake in the frontal cortex at 4 and 9 weeks).
Comparative PET, autoradiographic, and immunohistochemical assays of gliotic responses
Detailed maps of TSPO reflecting inflammatory responses to mechanistic injury at a regional/subregional scale were obtained by autoradiographic assays of postmortem brain slices with [18F]FE-DAA1106 (Fig. 5). Intensification of TSPO signals at the impact site became noticeable at 3 days, and peaked at 1 week. Moderately intense radiolabeling was also observed in the nearby neocortices and ipsilateral hippocampus. These radiolabelings diminished from 1 to 12 weeks, while radiosignals emerged at 8 to 12 weeks in the white matter connecting to the injury core, including the corpus callosum and external and internal capsules (bottom panels in Fig. 5).
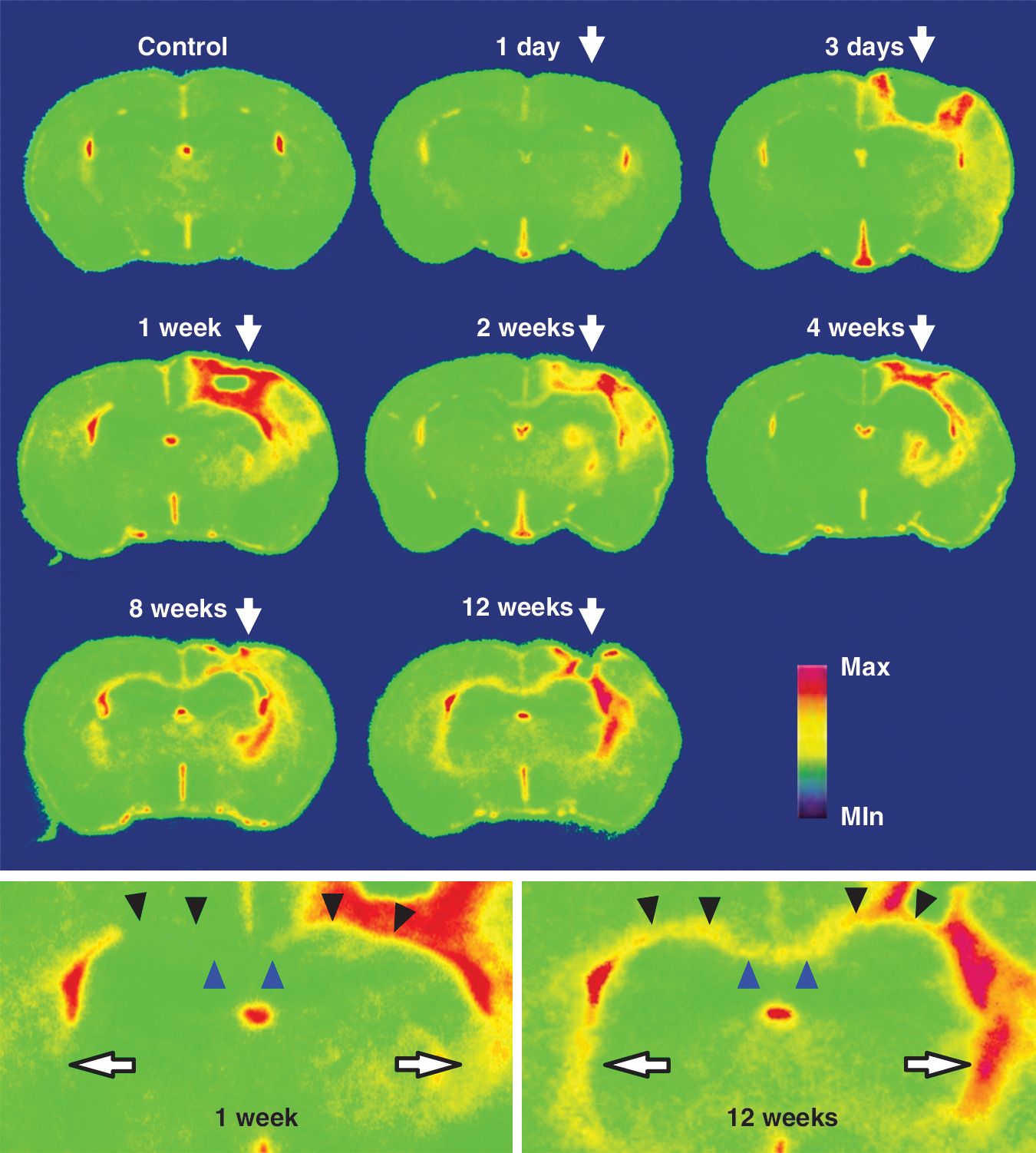
In vitro autoradiograms of [18F]fluoroethyl-DAA1106 in coronal rat brain slices containing the impact site (arrows in top panel) in a control animal and at different time points after traumatic injury. The slices were generated 2.5-mm posterior to the bregma. Magnified views of sections at 1 and 12 weeks are shown in the bottom panels. The blue and black arrowheads and white arrows in the bottom panels indicate the corpus callosum and external and internal capsules, respectively.
Brain sections were subsequently analyzed by immunofluorescence staining with antibodies against TSPO and microglial (Iba-1) and astrocytic (GFAP) markers (Fig. 6A). The appearance of TSPO signals concurrently with activation of microglia/macrophages and astrocytes was detectable in the proximity of the mechanical impact at 3 days. The intensities of Iba-1 and TSPO immunolabeling reached the highest levels at 1 week, consistent with PET and autoradiographic findings. During the period between 2 and 8 weeks, shrinkage of the necrotic lesion coincided with the reduction of TSPO-positive microglia/macrophages, while astrocytes doubly-positive for GFAP and TSPO encapsulating the inflammatory core remained abundant. Activation of TSPO-positive astrocytes was also recognizable in the bilateral external and internal capsules at 4 weeks, and became more prominent at 8 weeks. High-power microscopic examination revealed the emergence of TSPO-expressing microglia/macrophages at 3 days, followed by upregulation of TSPO in astrocytes forming a glial scar at 2–8 weeks, in the vicinity of the necrotic epicenter (Fig. 6B). Unlike the impact site, white matter distant from the mechanical injury exhibited minimal microgliotic changes, and activation of GFAP-immunoreactive astrocytes occurred at 1 week. TSPO signals were intensified in the vast majority of these cells at 4–8 weeks.

Immunofluorescence staining of coronal rat brain slices containing the impact surface (asterisks) at different time points after traumatic injury. (
Besides autoradiographic and immunohistochemical data, [18F]FE-DAA1106-PET images demonstrated increased TSPOs in the bilateral external and internal capsules in the late observation period (Fig. 7A). Quantitative indices of radiotracer uptakes, both uncorrected and corrected for body weights, indicated a biphasic elevation of TSPO levels, with their early (1 week) and delayed (4 weeks) rises being putatively attributable to microglial/macrophagic and astrocytic activations, respectively (Fig. 7B and C).
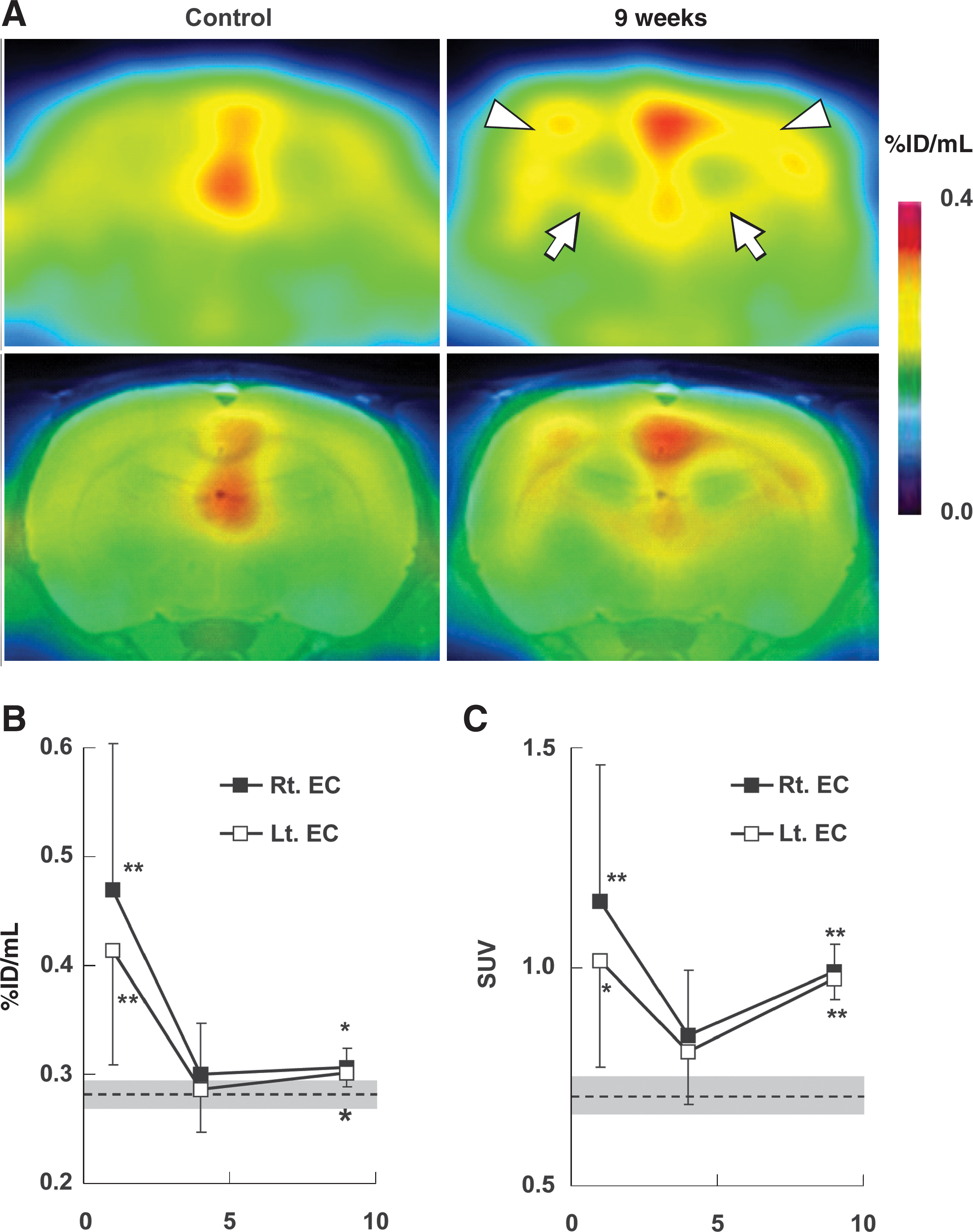
[18F]fluoroethyl-DAA1106 ([18F]FE-DAA1106) PET showing delayed enhancement of TSPO signals in the external and internal capsules. (
Discussion
The present longitudinal PET study with two distinct imaging agents covered the transition of TBI in rats from an acute to a chronic stage, and has documented the first evidence for a multiphasic alteration of the neuroinflammatory response and its interlinks to metabolic abnormalities. Although the severity of TBI may not be precisely defined due to the lack of phenotypic and neuropathological data, the observation that approximately a quarter of rats undergoing fluid percussion died of immediate cardiorespiratory arrest suggests a profound magnitude of the mechanical impact. In combination with autoradiographic and immunohistochemical assays, our in vivo image analyses indicated that a long-lasting intensification of TSPO signals in the proximity of the fluid percussion site arose from a relatively rapid accumulation of phagocytic microglia and macrophages at the necrotic core, followed by sustained encompassing of this primary inflammatory lesion by TSPO-expressing astrocytes. The population of TSPO-positive microglia and macrophages notably decreased along the shift from acute to subchronic inflammation between 1 and 4 weeks of injury, while a gradual upregulation of TSPO in astrocytes occurred during this period, and became more pronounced at the chronic phase beyond 8 weeks. Interestingly, astrogliotic changes were initiated at 1 week without noticeable expression of TSPO, and TSPO immunoreactivity emerged in this glial subspecies at 2–4 weeks, implying conversion of astrocytic functions from acute to chronic modes. These temporal sequences of gliotic responses were in good agreement with those in the ethanol-induced neurotoxicities determined by our previous study (Maeda et al., 2007a), but a more extensive TSPO rise was provoked by TBI in anatomical structures far beyond the impact boundary, including the contralateral frontal cortex, ipsilateral striatum, and bilateral subcortical white matter.
The striatum, one of the regions showing the lowest level of TSPO signals under physiological conditions, was involved in inflammatory responses to TBI, impeding the use of this region as a reference in quantitative analyses. Hence, interlateral asymmetry of radioactivities and indices for absolute radiotracer uptake requiring no reference tissues were applied to the determination of TSPO changes. Although contralateral TSPO elevation may considerably attenuate right-left contrasts in PET images, ipsilateral-to-contralateral ratio of in vivo TSPO radiolabeling served as an acceptably robust and sensitive index for probing inflammatory changes around the impact site, in light of the remarkable consistency between data from all 11 (Fig. 3C), and 6 selected (Fig. 3D), individual animals. Changes in other estimates, such as %ID/mL and SUV, could be confounded by the age of the animals, due to a notable growth in body mass disproportionate to the mild increase in brain weight seen over the observation period. This might lead to underestimation of TSPO levels by the use of %ID/mL at a late stage of TBI relative to an early phase. Similarly, additional increments of SUVs of TSPO radioligands observed from 4–9 weeks could result from overestimation of these values, since this rise was not consistent with diminished in vitro autoradiographic labeling during the period between 4 and 12 weeks. These considerations led to the notion that alterations of TSPO levels should be synthetically judged by a comparative use of indices sensitive and insensitive to body growth. Taking these in vivo indications together with immunostaining data, the multiphasicity of TSPO changes in the frontal cortex was accordingly summarized as a pronounced bilateral (but ipsilateral-dominant) surge by accumulation of TSPO-positive microglia and macrophages at 1 week, moderate ipsilateral elevation by concurrent microglial and astrocytic TSPO expressions at 4 weeks, and a modest bilateral rise primarily by astrocytic TSPO upregulation at 9 weeks.
There is also a methodological concern regarding the quantification of cerebral glucose utilization using [18F]FDG, as multiple lines of animal studies have documented the effects of anesthesia on the absolute uptake of this radiotracer and the spatial profile of its accumulation in the brain (Shimoji et al., 2004; Toyama et al., 2004). In fact, wide variability of %ID/mL values and SUVs was observed in the present PET measurements, presumably due to differences in depth of anesthesia among the experiments. Likewise, regional heterogeneities and target-to-cerebellum ratios of [18F]FDG radiosignals may be affected by the depth of anesthesia. In this context, the ipsilateral-to-contralateral ratio of radiotracer uptakes is useful as a reliable index for evaluating TBI-induced functional disruptions, despite the possibility of metabolic deficits in the contralateral structures. We here conceive that glucose consumption in the impacted frontal cortex was markedly disturbed at 1 week, and was marginally reduced at 4 weeks, based on ratios to the cerebellum (Fig. 3E) and contralateral cortex (Fig. 3F).
Inflammatory responses exhibited an overall correlation with metabolic abnormalities (Fig. 3A), while changes in right-left asymmetries of TSPO levels were more than threefold greater than those of glucose utilizations, supporting the use of TSPO imaging for sensitive detection of pathological alterations following head injuries. This is accounted for by low background signals of [18F]FE-DAA1106 in intact tissue, and isoflurane-induced metabolic suppression in normal regions may also decrease differences in [18F]FDG uptake between impacted and unimpacted sites. Moreover, it should be noted that both neuronal loss and astrocytic activation contribute to the cerebral glucose metabolism seen in a pathological condition. Glucose utilization for glutamate-glutamine cycling between neurons and astrocytes accounts for approximately 80% of the total glucose consumption in healthy brains (Magistretti et al., 1999; Shen et al., 1999), and high interstitial glutamate levels, as reported in TBI (Cavus et al., 2005; Richards et al., 2003), are likely to enhance glucose uptake into astrocytes (Magistretti and Pellerin, 1999). Hyperglycolytic states in activated astrocytes responding to acute to subchronic injuries could mask metabolic loss resulting from neuronal death at the impact site, and may plausibly explain the apparent recovery of interlateral [18F]FDG uptake ratios seen at 4 weeks regardless of the severity of acute metabolic deficits, which is in accord with previous clinical evidence (Bergsneider et al., 2001). Our observations also support the idea that high-level glucose consumption at 4 weeks indicates a large demand for astrocytic glycolysis due to profound neurotoxicity, and thus heralds persistent impairments of neuronal functionality at 9 weeks, with [18F]FDG uptake ratios being depressed concurrently with lowering astrocytic activities. However, application of this ratio at 4 weeks as a predictor of chronic deterioration might not be adequate, in light of the narrow range (within less than 4%) of the estimated values.
Since a close association of cerebral metabolic deficits with functional phenotypes has been reported in animal models of TBI (Dietrich et al., 1994; Ip et al., 2003; Moore et al., 2000), the coupling of the early inflammatory response and late metabolic diminution seen in these models may indicate the involvement of activated glia, which are putatively TSPO-positive microglia based on the present immunohistochemistry, in neuronal deterioration. This is in accord with our previous demonstration that TSPO-positive microgliosis emerges in a mouse model of neurodegeneration (Ji et al., 2008). As the implication of inflammatory gliosis in neurotoxicity was suggested in the same model by pharmacological immunosuppression (Yoshiyama et al., 2007), these findings imply a role of TSPO-positive microglia in the degradation of neuronal integrity. Meanwhile, whether this mechanistic concept is also applicable to TBI remains unclear, and might be determined by therapeutically intervening to suppress the neuroinflammation following impact. It should also be noted that the metabolic reduction may not necessarily be a direct indicator of neuronal deterioration, and thus contributions of TSPO-positive microglia to the loss of neuronal integrity would need to be assessed by neuropathological measures as well. In conducting such correlational in vivo and post-mortem assays, it should be considered that semi-quantitative indices, such as right-left asymmetry of radiotracer uptakes of [18F]FE-DAA1106 and [18F]FDG, might not be linearly proportional to each other and to the histopathological indices.
Detection of TSPO signals in white matter tracts at the chronic stage may be related to the emergence of TSPO-bearing astrocytes along axonal bundles. Unlike the proximity of the impact center, in vitro and in vivo TSPO positivity putatively derived from astrocytes was recognizable at relatively late phases of injury. An initial but transient rise of TSPO levels in white matter at 1 week was attributable to spillover of radiosignals from adjacent gray matter enriched with TSPO-expressing microglia and macrophages. The delayed upregulation of astrocytic TSPO could be a response to diffuse axonal injury, a common pathological feature of TBI (Smith et al., 2003; MacDonald et al., 2007). To obtain evidence more clearly supporting this notion, one would need to correlate TSPO radiosignals in the white matter with immunohistochemical indices of glial and axonal pathologies following impact, and to use therapeutic agents to counteract neuroinflammation.
To conclude, combined inflammatory and metabolic imaging represents a potentially powerful tool for clarifying the mechanistic involvement of glial cells and the disruption of metabolic homeostasis caused by neuron-astrocyte interactions seen during the progression from acute to chronic TBI. In consideration of the multiphasic gliotic and metabolic profiles revealed here, this methodology could also assist in the determination of optimal time frames for initiation of treatment to modulate neuronal and glial activity. TSPO-PET may provide a particularly useful imaging-based biomarker in therapeutic evaluations of anti-inflammatory and immunomodulatory agents, particularly in light of the accumulating experimental evidence of the efficacy of minocycline (Homsi et al., 2010; Sanchez Mejia et al., 2001) and cyclooxygenase inhibitors (Cernak et al., 2002; Browne et al., 2006) for the alleviation of post-traumatic neuropathology and functional deficits. Such pharmacological manipulations would also facilitate the clarification of the mechanistic roles played by TSPO-positive microglia and astrocytes at different stages post-TBI.
Footnotes
Acknowledgments
The authors thank Mr. Takeharu Minamihisamatsu for technical assistance, and the staffs at the Cyclotron Unit and the Molecular Probe Group for the cyclotron operation and production of radiochemicals. This work was supported in part by Grants-in-Aid for the Molecular Imaging Program and Scientific Research on Priority Areas, Research on Pathomechanisms of Brain Disorders 20023036 (to M.H.), and from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We also thank Taisho Pharmaceutical (Tokyo, Japan) for providing DAA1123.
Author Disclosure Statement
The authors declare no competing financial interests.