
Abstract
Heme oxygenase-1 (HO-1), a kind of stress protein, is critical for the protection against ischemic stroke and cerebrovascular endothelium damage. However, the effects of HO-1 on trauma-induced brain injury are still unknown. Hence, we attempted to use a cold injury-induced brain trauma (CIBT) model in mice, which provides for a well-established approach for assessing brain edema and blood–brain barrier breakdown. Additionally, we explored cultured mouse brain endothelial cells (bEnd.3) to investigate the protective effects of HO-1. HO-1 was induced by infection with a recombinant adenovirus carrying the human HO-1 gene or an inducer of HO-1 activity, cobalt protoporphyrin IX (CoPP). The recombinant adenovirus (3.5 × 107 PFU/mouse, i.v.) or CoPP (10 mg/kg, i.v.) significantly increased HO-1 protein expression and HO-1 enzyme activity in the cerebral cortex of the mice. We found that overexpression of HO-1 protected against cold injury-induced secondary damage and behavioral impairment. Up-regulation of HO-1 decreased brain edema and neutrophil infiltration induced by cold injury. These HO-1-dependent protecting effects were abrogated by pretreatment with the HO-1 inhibitor, zinc protoporphyrin IX (ZnPP; 3 mg/kg, i.v.). HO-1 expression in the cerebral endothelium was observed by immunofluorescent staining. CoPP-induced (1 μM, 24 h) HO-1 protein expression was determined by western blotting in bEnd.3 cells. Enhanced HO-1 also protected against cold injury-induced cell loss and damage, which were respectively determined by GAPDH leakage into the cell medium and XTT assay in bEnd.3 cells. In summary, HO-1 overexpression appears to offer an effective neuroprotection against cold-induced secondary brain injury.
Introduction
H
For brain injury, overexpression of HO-1 in transgenic mice effectively attenuated ischemic stroke damage produced by middle cerebral artery occlusion (Panahian et al., 1999). It is suggested that pharmacological stimulation of HO-1 activity may constitute a novel therapeutic approach in the amelioration of ischemic injury (Panahian et al., 1999). Until now, however, the effect of HO-1 on trauma-induced brain injury remains largely unknown.
Cold injury represents a common injury model for organ damage. Cold injury-induced brain trauma (CIBT) is a type of mixed injuries, including blood–brain barrier (BBB) breakdown, brain edema, neuronal apoptosis, and secondary lesion (Murakami et al., 1999; Nag et al., 1997). CIBT was attenuated in the superoxide dismutase (SOD)-1 transgenic (Tg) mice as compared to littermate non-Tg mice (Murakami et al., 1999). HO-1, as one of stress proteins, has been shown to protect rat livers from ischemia/reperfusion injury with extended cold preservation (Kato et al., 2001). An HO-1 byproduct, CO, has also been reported to protect against cold-rewarm-induced apoptosis (Stec et al., 2007). Whether HO-1 overexpression, like SOD-1, could protect against CIBT is still unknown and requires extensive study.
In this study, we used a CIBT model (Murakami et al., 1999) in mice to explore if HO-1 overexpression, induced by a recombinant adenovirus carrying the human HO-1 gene or a potent and effective inducer of HO-1 activity, CoPP, could rescue brain injury. We also used a mouse brain endothelial cell culture (bEnd.3) to investigate the cellular mechanisms of HO-1 protection on cold injury. Our results demonstrated that up-regulation of HO-1 in cerebral vascular endothelium protected cold injury-induced brain and cellular damage in mice.
Methods
Materials
The polyclonal antibodies to HO-1 and myeloperoxidase (MPO) were from Santa Cruz (Santa Cruz, CA). The antibody to Von Willebrand Factor was from DAKO (A/S, Denmark). The anti-GAPDH antibody was from Biogenesis (Bournemouth, UK). 2′,7′-Dichlorofluorescein diacetate (DCF-DA) was from Molecular Probes (Eugene, OR). Bicinchoninic acid (BCA) protein assay reagent was from Pierce (Rockford, IL). CoPP, biliverdin reductase, tetrazolium salt XTT reagent, and other chemicals were from Sigma (St. Louis, MO).
Study groups and experimental protocol
Male ICR mice (6–8 weeks old and weighing 30–34 g) were obtained from the Laboratory Animal Center of National Taiwan University (Taipei, Taiwan). The mice were housed indoors in the Laboratory of Animal Resource Center at Chang Gung University under automatically controlled temperature and light cycle, and were fed standard laboratory chow and tap water ad libitum. The procedures used in this study were reviewed and approved by the Animal Use Committee of Chang Gung University as fulfilling the institutional guidelines for animal use.
To investigate the role of HO-1 in CIBT, a recombinant adenovirus carrying the human HO-1 gene, a HO-1 inducer, cobalt protoporphyrin IX (CoPP), and a HO-1 inhibitor, zinc protoporphyrin IX (ZnPP), were used in the present study. The mice were randomly divided into ten groups: (a) sham control mice (mice were sham injured, n = 6); (b) recombinant adenovirus-treated group (Adv, mice were tail-vein injected with the adenovirus on days 1, 3, 5, and 7, 3.5 × 107 PFU/each injection/mouse, n = 6); (c) recombinant adenovirus carrying the human HO-1 gene-treated group (Adv-HO-1, mice were tail-vein injected with HO-1 gene containing adenovirus on days 1, 3, 5, and 7, 3.5 × 107 PFU/each injection/mouse, n = 6); (d) CoPP-treated group (CoPP, mice were tail-vein injected with CoPP for 24 h, n = 8); (e) ZnPP-treated group (ZnPP, mice were tail-vein injected with ZnPP for 2 h, n = 8); (f ) cold injury group (CI, n = 8); (g) AdV-pretreated group (AdV/CI, mice were tail-vein injected with adenovirus on days 1, 3, 5, and 7 before cold injury, n = 6); (h) AdV-HO-1-pretreated group (AdV-HO-1/CI, mice were tail-vein injected with HO-1 gene containing adenovirus on days 1, 3, 5, and 7 before cold injury, n = 6); (i) CoPP-pretreated group (CoPP/CI, mice were tail-vein injected with CoPP for 24 h before cold injury, n = 8); and (j) CoPP-ZnPP-pretreated group (CoPP-ZnPP/CI, mice were tail-vein injected with CoPP for 24 h before cold injury and ZnPP for 2 h before cold injury, n = 8). The number of animals used in each experimental group was between six and eight.
Cold injury-induced mice brain trauma model
CIBT was applied as previously described (Murakami et al., 1999), with slight modifications. The mice were anesthetized with Pentothal (0.3 ml/kg, i.p.) and deep anesthesia was maintained by inhalation of ether during the period of cold injury. The scalp was incised on the midline, and the skull was exposed. A defined area of the right parietal bone (2 mm behind bregma, 2 mm lateral of the sagittal suture; diameter 3 mm) was then thinned to translucency. An iron probe of 100 g with a 3-mm diameter, cooled with liquid nitrogen, was applied to the thinned skull area for 30 sec. After trauma induction, the skin incision was sutured, and the animals were allowed to wake up fully before experimentation.
Behavior tests
The neurological evaluation was performed as previously described (Merkler et al., 2001), with some modifications. Behavioral tests were performed at 24 h after sham injury or CIBT. In brief, the plane balancing test was used to assess the ability of the mice to balance on a platform 5 cm in diameter and 50 cm in height. The mice were trained at least three times to learn to stay on the platform for more than 60 sec before the cold injury experiment. The resting time of each mouse that stayed on the platform was scored three times. The beam walk test was used to assess the ability of mice to balance on 5-cm elevated glassy beams with a round cross-section (2.5 cm in diameter) with a length of 30 cm and a height of 50 cm. The mice were trained at least three times to learn to walk from one end to the other end without falling down before the cold injury experiment. Each mouse was tested on the beam walk test 10 times. The rate of successive walking from one end to the other end was calculated as the score of the beam walk test.
Assessment of blood–brain barrier breakdown and vasogenic brain edema
Evans blue dye extravasation was used to quantify the BBB damage (Cao et al., 2006). After the behavior test, Evans blue (3%, dissolved in phosphate-buffered solution [PBS]) was injected (0.2 ml/mouse, i.p.) for 2 h before sacrifice. Deep anesthesia was induced by injection of pentothal (0.05 ml/mouse, i.p.) and maintained by inhalation of ether. The mice were perfused with ice-cold PBS. After decapitation, the brains were quickly removed and pictures were taken. The area stained with Evans blue dye extravasation on the surface of cerebral cortex was quantified by the Axiovert 200M system (Carl Zeiss, Germany). For vasogenic brain edema assessment, the mice were deeply anesthetized and immediately decapitated after the behavior test without the perfusion process (Paul et al., 2007). The brains were quickly removed and divided into three parts: ipsilateral cortex, contralateral cortex, and cerebellum (which served as an internal control). Brain samples were immediately weighed to obtain the wet weight, and were dried at 60°C for 120 h to obtain the dry weight. Brain edema was expressed as (wet weight – dry weight)/wet weight of brain tissue × 100%. There were at least six mice per group.
Preparation of recombinant adenovirus
A recombinant adenovirus containing human HO-1 (Adv-HO-1) was provided by Dr. L.Y. Chau (Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan). Recombinant adenovirus was generated by homologous recombination and amplified in HEK-293 cells (transformed primary embryonic kidney, human, ATCC CRL 1573). The same batch of viral vectors was purified by CsCl ultracentrifugation and stored in 10 mM Tris-HCl (pH 7.4), 1 mM MgCl2, and 10% (v/v) glycerol at −80°C until used for experiments. Virus titers were determined by a plaque assay on a HEK-293 cell monolayer. Mice were tail-vein injected (3.5 × 107 PFU/mouse) with recombinant adenovirus carrying the human HO-1 gene on days 1, 3, 5, and 7 before the cold injury experiment.
HO-1 immunofluorescent staining
The mice were deeply anesthetized and perfused with ice-cold PBS followed by 4% paraformaldehyde. After decapitation, the brains were quickly removed, soaked in 30% sucrose medium overnight, frozen with liquid nitrogen, and then stored at −80°C.
For immunofluorescent staining, cryostat coronal brain sections (15-μm thickness) were incubated with a goat anti-HO-1 antibody (1:400 dilution, at 4°C for 24 h), as well as rabbit anti-Von Willebrand Factor polyclonal antibody (1:400 dilution, at 4°C for 24 h), an endothelial cell marker, for double immunostaining. Brain sections were then incubated with a fluorescein isothiocyanate (FITC)-conjugated anti-goat antibody and rhodamine-conjugated anti-rabbit antibody (1:100 dilution, at room temperature for 1 h). Slices were mounted with aqueous mounting medium to image under a ZEISS microscope equipped with an Axiovert 200M system (Carl Zeiss).
Enzymatic assay for HO-1 activity
HO activity was measured based on the levels of bilirubin formation using microsomal fraction of cells as source (Tenhunen et al., 1971). Briefly, mice cerebral cortex or bEnd.3 cells were homogenized and sonicated before centrifuged at 15000g for 40 min at 4°C. The supernatant (500 μg of protein) was mixed with NADH-containing buffer (containing 1 mM NADH, 2 mM glucose 6-phosphate, 2 U glucose-1-phosphate dehydrogenase, 600 μg of biliverdin reductase, 1 mM potassium phosphate buffer, and 25 μM hemin) for 60 min at 37°C in the dark. The reaction without the NADH served as a blank. The reactions were stopped by placing the mixture on ice, subsequently scanned with a spectrophotometer. The amount of bilirubin was determined by the difference in optical density units between 470 and 550 nm.
Determination of leukocyte infiltration
Leukocyte infiltration was evaluated as described previously (Wang and Doré, 2007). In brief, the coronal brain sections were prepared as described in above. Sections of damaged brain area choose from different groups were incubated with an anti-myeloperoxidase antibody (MPO, 1:200 dilution at 4°C for 24 h), a specific marker for microglia/macrophages, followed by an FITC-conjugated secondary antibody. The images of stained sections were captured with a ZEISS microscope equipped with an Axiovert 200M system (Carl Zeiss).
Cell cultures
Mouse brain endothelial cells (bEnd.3, ATCC CRL-2299) were grown in DMEM/F-12 containing 10% FBS and antibiotics (100 U/ml penicillin G, 100 μg/ml streptomycin, and 250 ng/ml fungizone) at 37°C in a humidified 5% CO2 atmosphere. When the cultures grew to confluence (about 4 days), cells were detached with 0.05% (w/v) trypsin/0.53 mM EDTA for 5 min at 37°C. The cell suspension was diluted with DMEM/F-12 containing 10% FBS to a concentration of 2 × 105 cells/ml. The cell suspension was plated onto 12-well culture plates (1 ml/well) for the measurement of protein expression. The culture medium was changed after 24 h and every 3 days. Experiments were performed with cells from passages 5 to 13.
In vitro cold ring injury
bEnd.3 cells were cultured onto a 3-cm culture dish (2 ml/dish). Cold ring injury (CRI) was performed with an iron ring (outer diameter 25 mm, inner diameter 10 mm), which was precooled with liquid nitrogen. The cold ring was placed in contact with the outside of the culture dish bottom for 60 sec. The culture dishes were then transferred into a CO2 incubator for 24 h. Cold ring damaged cells and these cells detached from the cultured dish and released into cultured medium. CRI-induced cellular damage was measured by detecting a housekeeping protein, GAPDH, in cell lysate and the cultured medium using western blotting.
In vitro cytotoxicity assay
The XTT method of monitoring in vitro cytotoxicity was applied according to the manufacturer's manual (tetrazolium salt XTT reagent, TOX-2; Sigma). In brief, bEnd.3 cells were cultured onto 3-cm culture dishes (2 ml/dish). When the cultures grew to 90% confluence, cells were serum deprived for 24 h in RPMI 1640 medium (phenol red free) prior to cold ring injury. Twenty-four hours after the cold ring injury, XTT was added into the culture medium with a final concentration of 20% for 2 h. The culture medium was measured by spectrophotometric absorbance at a wavelength of 450 nm.
Cell extracts preparation and western blot
Western blot analysis was performed as described previously by Lee and associates (2004). For brain tissue, mouse brains were homogenized in a lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS (pH 8.0), then centrifuged at 13,000g for 30 min. The protein concentration of the supernatant was measured using a BCA reagent. For cell cultures, bEnd.3 cells were lysed with a sample buffer containing 125 mM Tris-BASE, 5% glycerol, 3% β-mercaptoethanol, 1.25% SDS, and 0.05% bromophenol blue. Equal amounts (30 μg) of total protein were denatured, subjected to SDS-PAGE using a 12% running gel, and transferred onto a nitrocellulose membrane. Membranes were incubated with a goat anti-HO-1 antibody (1:1000 dilution at 4°C for 24 h), and then incubated with an anti-goat horseradish peroxidase antibody (1:2000 dilution, at room temperature for 1 h). The gel bands detected by ECL reagents were developed by Hyperfilm-ECL and were quantified by a densitometry.
Statistical analysis
Values were expressed as means ± SEM. Statistical evaluation was performed using the Student's t test and one-way analysis of variance (ANOVA) followed by post hoc Dunnett's test. A p value of < 0.05 was considered significant.
Results
HO-1 expression by a recombinant adenovirus protects against cold injury-induced brain secondary damage and behavioral impairments
As shown in Figure 1, HO-1 expression (Fig. 1A) and enzyme activity (Fig. 1B) were significantly increased in the cerebral cortex tissues of mice infected with a recombinant adenovirus carrying human HO-1 gene. Cold injury induced an increase in the damaged area of cerebral cortex at 4 h and 24 h (Fig. 1C). HO-1 overexpression by a recombinant adenovirus (Adv-HO-1) had no effect on brain damage caused by immediate physical injury within 4 h, but could improve cold injury-induced secondary damage within 24 h (Fig. 1C and D). Cold injury induced a decreased score in both the plane balancing test (Fig. 1E) and the beam walk test (Fig. 1F) at 24 h. HO-1 overexpression restored the scores in both the plane balancing test (Fig. 2E) and the beam walk test (Fig. 2F) compared with those of the cold injury group. HO-1 gene overexpression may offer a protective role in cold injury-induced cerebral cortex secondary damage and neurological/behavioral impairment.

HO-1 expression by a recombinant adenovirus HO-1 gene protects against cold injury-induced brain trauma. (
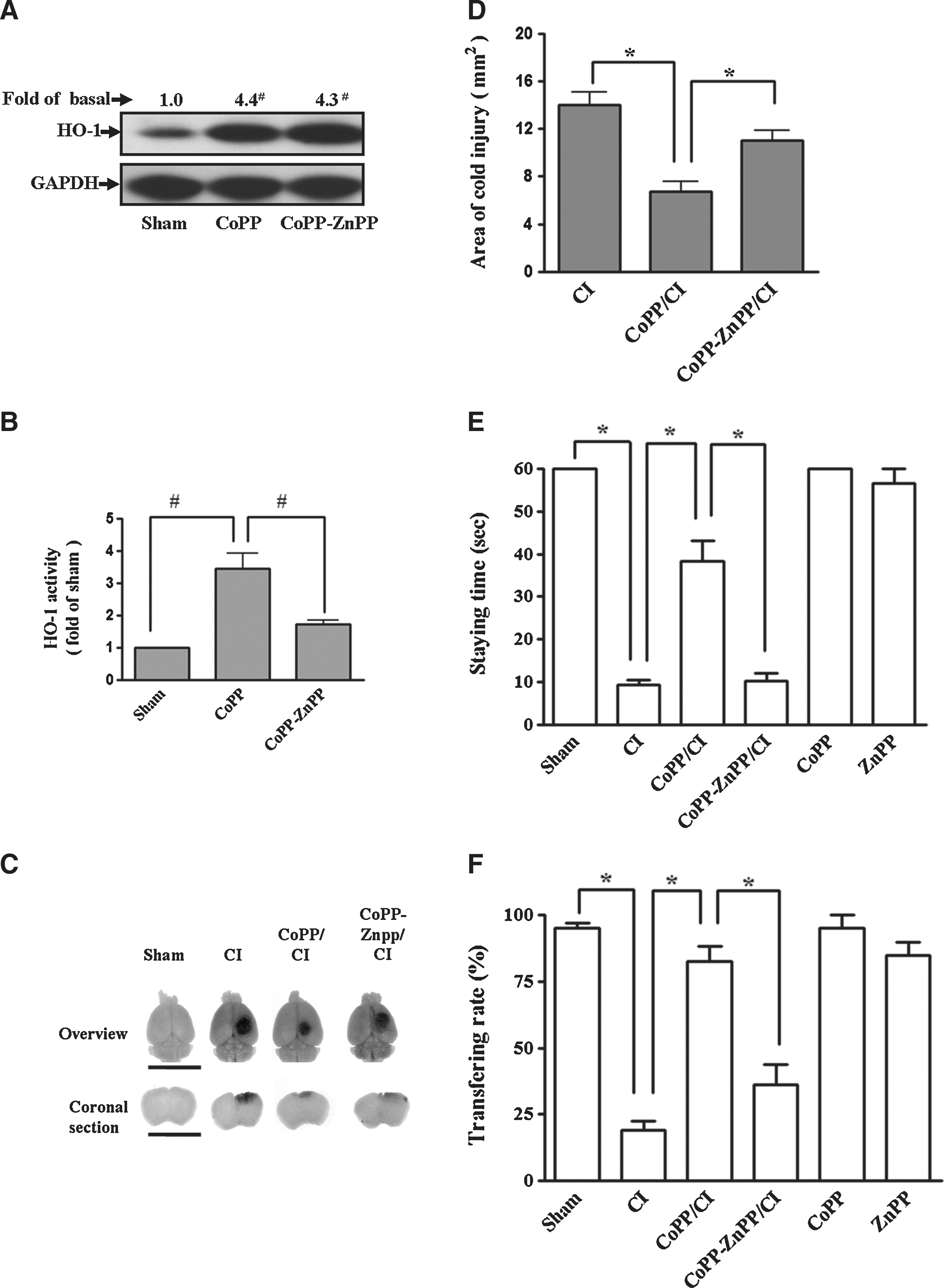
HO-1 expression by a pharmacological inducer protected against cold injury-induced brain trauma. (
HO-1 expression by a pharmacological inducer protects against cold injury-induced brain cerebral cortex damage and neurological/behavioral impairments
As shown in Figure 2, CoPP (10 mg/kg, 24 h, tail-vein injection) treatment significantly increased HO-1 protein expression (Fig. 2A) and enzyme activity (Fig. 2B) in cerebral cortex. Cold injury induced an increase in the damaged area of cerebral cortex (Fig. 2C) and a decreased score in both the plane balancing test (Fig. 2E) and the beam walk test (Fig. 2F) at 24 h. Pretreatment with CoPP for 24 h reduced the area of brain damage (Fig. 2C and D) and restored the scores in the neurological/behavioral tests as compared to those of the cold injury group (Fig. 2E and F). These data suggest that pharmacological-induced HO-1 may offer a protective role in cold injury-induced cerebral cortex damage and neurological/behavioral impairment.
To ascertain that the protective effect of CoPP on cold injury-induced cerebral cortex damage was associated with HO-1 expression, the mice were pretreated with ZnPP (3 mg/kg, i.p.) 2 h before cold injury. ZnPP treatment had no effect on CoPP-induced HO-1 protein expression (Fig. 2A) but significantly decreased HO-1 enzyme activity (Fig. 2B). ZnPP effectively reversed the protective effects of CoPP on cold injury-induced brain cerebral cortex damage (Fig. 2C and D) and neurological impairment (Fig. 2E and F).
Up-regulation of HO-1 decreases brain edema and neutrophil infiltration induced by cold injury
Cold injury also increased the water content in the ipsilateral cortex (Fig. 3A). Pretreatment with CoPP significantly reduced cold injury-induced brain edema. On the other hand, ZnPP treatment reversed the CoPP-produced neuroprotection on brain edema, suggests that HO-1 could play a critical role in preventing brain edema. As shown in Figure 3B, there were more infiltrating neutrophils around the injured area (IA) in cold injury treated mice than those of the control group, detected by immunofluorescence staining using an anti-MPO antibody. Pretreatment with CoPP effectively decreased the cold injury-induced neutrophil infiltration (Fig. 3B).

HO-1 pharmacological inducer decreased cold injury-induced brain edema and neutrophils infiltration. (
HO-1 protected cold injury-induced brain damage via cerebral endothelial cells
HO-1 expression was found to be colocalized with cerebral endothelial cells detected by anti-HO-1 and anti-Von Willebrand Factor double immunofluorescent staining (Fig. 4). As shown in Figure 5A and B, treatment with CoPP (1 μM) for 24 h induced HO-1 protein expression in mouse brain endothelial cells (bEnd.3). Cold ring injury (CRI, 24 h) damaged bEnd.3 cells, which reflected as a significant loss of housekeeping protein GAPDH in cell lysates detecting by a leaked GAPDH, appeared in the cultured medium (Fig. 5A and B). Pretreatment with CoPP induced HO-1 protein expression and protected cellular damage in bEnd.3 cells (Fig. 5A and B). Moreover, the data obtained with XTT assay showed that CRI decreased cell viability by approximately 40% at 24 h after onset (Fig. 5C). Pretreatment with CoPP (1 μM, 24 h) attenuated CRI-induced cellular damage, whereas pretreatment with ZnPP (0.1 μM, 1 h before cold ring injury) abrogated this cytoprotective effect (Fig. 5C). These results indicated that HO-1 prevented the development of cold injury-induced damage in mouse brain endothelial cells.

Expression of HO-1 in mouse cerebral endothelial cells. Extended alterations of HO-1 expression, in cortex, colocalized with cerebral microvessels confirmed by an immunofluorescent double staining using an HO-1 antibody (green) and an endothelial cell marker (Von Willebrand Factor, red; phase contrast × 200, scale bar = 30 μm).

HO-1 protein expression protects against cold ring injury-induced cellular damage in bEnd.3 cells. (
Discussion
HO-1, a kind of stress protein, is critical for protection from ischemic stroke and cerebrovascular endothelium damage. Overexpression of HO-1 has been shown to attenuate ischemic stroke induced by middle cerebral artery occlusion (Panahian et al., 1999). In addition, it has also been reported that using HO-1 transgenic mice as a model to evaluate neuroprotection demonstrated that HO-1 exerts as a brain cellular defense mechanism (Maines, 2002). Vascular induction of HO-1 by heme was reported to stabilize the blood–spinal cord barrier and limit early leukocyte infiltration in the injured spinal cord (Lin et al., 2006; Yamauchi et al., 2004). Moreover, in the present study, cold injury-induced brain trauma was proved to be reduced by an up-regulation of HO-1 expression.
Many acute neurological diseases, such as brain trauma and stroke, are related to vasogenic brain edema and secondary brain lesion (Blight, 2002; Paul et al., 2007), where BBB damage and oxidative stress play a key role in these pathogenic consequences (Grzeschik et al., 2003; Jones et al., 2005; Murakami et al., 1999). Cerebral endothelial disruption induced by oxidative stress (Ryter and Choi, 2005; Yachie et al., 1999) or excitotoxicity (Parfenova et al., 2006) was diminished by treatment of stress protein HOs, especially HO-1, suggesting that HO-1 overexpression contributes to cerebral microvascular protection in brain injury. HO-1 represents an important endogenous antioxidative defense mechanism against tissue damage (Ryter and Choi, 2005). Moreover, in this study, HO-1 overexpression induced by a recombinant adenovirus-HO-1 or CoPP protects against cold injury-induced brain secondary damage and behavioral impairments. We also found that up-regulation of HO-1 decreased brain edema and neutrophil infiltration induced by cold injury. Taken together, these results suggest that cerebral vascular induction of HO-1 expression could protect cold injury-induced BBB damage. On the other hand, HO-1 gene overexpression has been shown to attenuate ischemic stroke induced by middle cerebral artery occlusion (Panahian et al., 1999). However, the injury volume of intracerebral hemorrhage was significantly smaller in HO-1 knockout (HO-1−/−) mice than those of wild-type controls, suggesting that HO-1 might conversely exacerbate the intracerebral haemorrhagic injury (Wang and Doré, 2007). Since pathological conditions are quite different between brain ischemia and hemorrhage, HO-1 may play different roles under these two types of stroke.
Cold injury-induced oxidative stress is known to enhance early secondary tissue damage (Murakami et al., 1999). Superoxide dismutase (SOD)-1 transgenic mice with overexpressed copper, zinc-SOD decreased CIBT, suggesting that oxygen radicals, superoxide anion in particular, mediate the development of CIBT (Murakami et al., 1999). However, there is no significant difference in CIBT between wild type and manganese-SOD (MnSOD, SOD2) knockout mice, suggesting that the accumulation of free radicals may not play a significant role in secondary brain damage. Interestingly, there are differential expressions between MnSOD and HO-1 proteins in cerebral endothelial cells in response to sublethal oxidative stress (Méthy et al., 2004). HO-1expression is more sensitive than MnSOD in response to oxidative stress. The role of oxidative stress in the development of secondary brain damage needs more detailed investigation. Cold injury is a complex type of organ injury. In the present study, HO-1, as one member of anti-oxidative stress protein, reduced cold injury-induced secondary brain damage 24 h, not 4 h, after cold injury (Fig. 1B), suggesting that HO-1 overexpression could prevent secondary brain damage within 4–24 h after cold injury.
HO-1 plays an important role during repair processes after cold injury because HO-1 limits secondary brain damage after mouse brain cold injury model in the present study. For the same model, it has been also reported that astrocyte derived-type VIII collagen may play an important role after cold injury, because it was up-regulated around the necrotic region during the repair process (Hirano et al., 2004). Since HO-1 converts heme from a prooxidant into antioxidant molecules, the induction of HO-1 would be expected to augment the oxidative defense mechanisms compromised by brain injury (Nimura et al., 1996). The protection of HO-1 may be contributed from the byproduct, CO, which has been reported to protect against cold-rewarm-induced apoptosis (Stec et al., 2007).
Several lines of evidence have demonstrated a protective role of HO-1 expression on brain injury. First, HO-1 plays as a modulator for several growth factors, that is, vascular endothelial growth factor-A, the transforming growth factors family, hepatocyte growth factor/scatter factor, platelet-derived growth factor, fibroblast growth factor, and nerve growth factors (Hill-Kapturczak et al., 2007). Second, HO-1 could be linked to action of stromal-derived factor-1, a crucial chemokine responsible for attraction of progenitor cells to the site of ischemia (Deshane et al., 2007). HO-1 protected endothelial cells against not only heme but also different inflammatory mediators, preventing the apoptosis but stimulating proliferation (Soares et al., 2002). However, up to now, pharmacological inducers that could trigger HO-1 overexpression to provide neuroprotection against brain damage are not known. Our current finding is the first time pharmacological compound(s) have been identified that can enhance HO-1 overexpression and activity in mice cerebral vascular endothelial cells, in vivo and in vitro, consequently protecting against CIBT. These results create opportunities for the development of preventive strategies on various types of brain damage. On the other hand, Bergeron and associates (1998) have demonstrated that little or no basal HO-1 expression was presented in endothelium and cortex in adult rats. Consistently, we also observed fewer HO-1 positive cells in the cerebral cortex of control mice (data not shown). In addition, HO-1 has been noted to be markedly induced by various insults in the central nervous system (Schipper, 2004). However, in the present study, although cold injury caused severe brain damage, no change in levels of HO-1 protein was observed (data not shown). These results suggested that HO-1 expression is not affected by cold injury, but drug-induced HO-1 up-regulation could effectively protect the cold injury-induced brain from secondary damage and neurological impairments.
As with the properties of tightly binding to albumin, Evans blue dye is widely used as a tracer of plasma to investigate cellular membrane permeability, especially endothelial cells (Patterson et al., 1992). Evans blue dye does not pass the BBB into the brain tissue. Only when there is barrier disruption does Evans blue dye coexist with leakage plasma in the brain tissue. Although the level of Evans blue in the bloodstream could account for staining of the damaged cortex, there is no direct evidence that increased HO-1 levels could affect the uptake of Evans blue from the peritoneal cavity (where it was injected in the present study) into the bloodstream. Therefore, up-regulation of HO-1 decreased staining of Evans blue on cortex was due to its protecting effect against cold injury-induced barrier disruption.
In addition, CoPP, a non-substrate HO-1 inducer, has been reported to protect the endothelial injury (Ewing et al., 2005; Johns et al., 2009). The effective doses of intraperitoneally injected CoPP in animal studies were around 5–20 mg/kg (Johns et al., 2009; L'Abbate et al., 2007). In this study, CoPP at 10 mg/kg has been shown to up-regulate HO-1 protein expression and enzyme activity in mice. In contrast, CoPP-induced neuroprotection could be reversed by pretreatment with HO-1 activity inhibitor, ZnPP, confirmed the predominant effect of HO-1 activation by CoPP manipulation.
In conclusion, we demonstrated that HO-1 gene overexpression or pretreatment with pharmacological inducer CoPP protected against CIBT. These protective effects may be mediated through HO-1 expression in cerebral endothelium. HO-1 may play an important role in the repair process after cold injury. The pharmacological stimulation of HO-1 expression may offer a novel clinical strategy in amelioration of brain trauma.
Footnotes
Acknowledgments
This work was supported by National Science Council, Taiwan, grant numbers NSC97-2321-B-182-007, NSC98-2321-B-182-004, and NSC96-2320-B-182-009; and Chang Gung Medical Research Foundation, grant numbers CMRPD150253, CMRPD150313, CMRPD170492, and CMRPD180371.
Author Disclosure Statement
No competing financial interests exist.