
Abstract
To elucidate the putative neuroprotective effects of ghrelin in subarachnoid hemorrhage (SAH)-induced brain injury, Wistar albino rats (n = 54) were divided into sham-operated control, saline-treated SAH, and ghrelin-treated (10 μg/kg/d IP) SAH groups. The rats were injected with blood (0.3 mL) into the cisterna magna to induce SAH, and were sacrificed 48 h after the neurological examination scores were recorded. In plasma samples, neuron-specific enolase (NSE), S-100β protein, TNF-α, and IL-1β levels were evaluated, while forebrain tissue samples were taken for the measurement of malondialdehyde (MDA), glutathione (GSH), reactive oxygen species levels, myeloperoxidase (MPO), Na+-K+-ATPase activity, and DNA fragmentation ratio. Brain tissue samples containing the basilar arteries were obtained for histological examination, while cerebrum and cerebellum were removed for the measurement of blood–brain barrier (BBB) permeability and brain water content. The neurological scores were impaired at 48 h after SAH induction, and SAH caused significant decreases in brain GSH content and Na+-K+-ATPase activity, and increases in chemiluminescence, MDA levels, and MPO activity. Compared with the control group, the protein levels of NSE, S-100β, TNF-α, and IL-1β in plasma were also increased, while ghrelin treatment prevented all SAH-induced alterations observed both biochemically and histopathologically. The results demonstrate that ghrelin alleviates SAH-induced oxidative brain damage, and exerts neuroprotection by maintaining a balance in oxidant-antioxidant status, by inhibiting proinflammatory mediators, and preventing the depletion of endogenous antioxidants evoked by SAH.
Introduction
Ghrelin is a 28-amino acid peptide that has been discovered in rat and human gastrointestinal tract, particularly in gastric mucosa, as an endogenous ligand for growth hormone secretagogue receptor (GHS-R; Kojima et al., 1999). Ghrelin is secreted in substantial amounts into the circulation by the stomach and the intestine, while the central nervous system (CNS), kidneys, placenta, pancreas, and pituitary all produce small amounts of ghrelin (Horvath et al., 2001). In addition to its well-described effects on appetite regulation (Cummings et al., 2002) ghrelin has been shown to have anti-inflammatory properties. We have recently shown that ghrelin alleviates pancreatic and hepatic injury, and improves burn-induced multiple organ injury by depressing neutrophil infiltration and the release of pro-inflammatory cytokines (Iseri et al., 2008; Kasımay et al., 2006; Sehirli et al., 2008). Recently, ghrelin was also shown to exert neuroprotective effects in several models of neuronal injury (Liu et al., 2006; Miao, 2007; Obay et al., 2008; Xu et al., 2009).
Considering that oxidative stress plays an important role in the pathogenesis of acute brain injury after SAH (Pyne-Geithman et al., 2009; Sano et al., 1980), reducing oxidative stress could be predicted as a therapeutic target for acute brain injury after SAH. Accordingly, the use of some free radical scavengers in experimental models (Erşahin et al., 2009; Imperatore et al., 2000; Ostrowski et al., 2006), and in some clinical trials (Munakata et al., 2009), has proven to be beneficial in the treatment of aneurysmal SAH. In light of the aforementioned studies, in the present study we aimed to determine the possible anti-inflammatory effects of ghrelin on SAH-induced oxidative brain injury and neurological symptoms using biochemical, neurological, and histopathological approaches.
Methods
Animals and experimental design
All experimental protocols were approved by the Marmara University Animal Care and Use Committee. Male Wistar albino rats (300–350 g) were housed in a temperature-controlled room (22 ± 2°C) with a standardized light/dark cycle (12/12 h), and the relative humidity (65–70%) was kept constant. The rats were fed with standard rat pellets and tap water ad libitum.
The rats were randomly divided into three groups: a control group (sham-operated and saline-treated; n = 18), a SAH group (SAH-operated and saline-treated; n = 18), and a ghrelin + SAH group (SAH-operated and ghrelin-treated 10 μg/kg/d IP; n = 18). Either ghrelin or saline was administered immediately after the surgery and was repeated 24 h later. Three hours after surgery, blood samples were withdrawn from the tail vein to study the plasma levels of cytokines and the markers of brain injury. At the 48 h post-surgery, neurological examinations were performed in all groups. Then randomly selected rats in each group were decapitated to obtain trunk blood and brain tissue samples for biochemical analysis (n = 6), and for Evans blue assay and edema evaluation (n = 6). The remaining rats (n = 6) in each group were given fixative perfusion for histological preparation and analysis. Forebrain tissue samples were taken for the measurement of malondialdehyde (MDA), glutathione (GSH), and reactive oxygen species levels, myeloperoxidase (MPO) and Na+-K+-ATPase activity, and DNA fragmentation ratio. Brain tissue samples containing the basilar arteries were obtained for histological examination, while cerebrum and cerebellum were removed for the measurement of blood–brain barrier (BBB) permeability and brain water content.
Assessment of memory function
To evaluate the short-term effect of SAH injury on memory function, passive avoidance testing was carried out using a box (Northel Passive Avoidance System, İstanbul, Turkey) composed of an illuminated (with a 100W bulb) and a non-illuminated compartment (each 20 × 20 × 20 cm). The two compartments were separated by a guillotine door (5 × 5 cm), and the floor of the dark compartment had 2-mm stainless steel rods spaced 1 cm apart. For the acquisition trial performed 24 h prior to sham- or SAH-surgery, the rats were initially placed in the illuminated compartment, and the door between the two compartments was opened 10 sec later. When the rats entered the dark compartment, the door automatically closed and an electrical foot shock (0.3–0.6 mA) that lasted for 5 sec was delivered through the stainless steel rods. Forty-eight hours after surgery (3 days after the acquisition trial), the rats were again placed in the illuminated compartment for the retention trials. The time taken for a rat to enter the dark compartment after door opening was measured as the latency time in both the acquisition and retention trials. If the rat did not enter the dark compartment within 240 sec, it was assumed that the rat remembered the single training trial.
Induction of subarachnoid hemorrhage
In the present study, a previously described model of basilar artery vasospasm was used (Delgado et al., 1985). In the spontaneously breathing anesthetized rat injected IP with ketamine (100 mg/kg) and chlorpromazine (2 mg/kg), a small incision was made in the area of the occipitocervical junction to expose the atlanto-occipital membrane. After the animal was placed in the stereotaxic frame, the cisterna magna was tapped with a 27-gauge needle, and 0.3 mL of cerebrospinal fluid was gently aspirated. Freshly drawn blood (0.3 mL) taken from the femoral artery was then injected aseptically into the cisterna magna within a 2-min period. Immediately after the injection of blood, the puncture site was sealed with glue to prevent the formation of a fistula. To permit blood distribution around the basal arteries, each rat was tilted at an angle of 20° for 30 min with its head lowered.
Neurological examination
Since functional scoring is important in testing neuroprotective drugs, a simple set of commonly used neurological tests was used to assess normal and abnormal function following SAH in rats. The neurological examinations were conducted according to Bederson's modified neurological examination test (Toklu et al., 2009a) by a blinded investigator. A 20-point neuroscore was used to assess motor and behavioral deficits. The sequence of testing animals by any given task was randomized. Briefly, the consciousness, performance in a smooth climbing platform, extremity tonus, walking and postural reflexes, circling, and response to the nociceptive stimuli were assessed. For walking and posture, the rats were allowed to move about freely on the floor while they were observed. In the circling test, the rats were held gently by the tail, suspended one meter above the floor, and observed for forelimb flexion, for which normal rats are expected to extend both forelimbs toward the floor. The rotation degree and time were measured. Finally, the responses to nociceptive stimuli were assessed by tail-immersion testing in 56°C water.
Blood assays
Plasma levels of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) were quantified using enzyme-linked immunosorbent assay (ELISA) kits specific for the previously mentioned rat cytokines according to the manufacturers' instructions and guidelines (Biosource Europe S.A., Nivelles, Belgium). Plasma levels of neuron-specific enolase (NSE), and soluble protein-100β (S-100β), indices of neuron and astrocyte injury, respectively, were assayed using ELISA kits for rats (USCN Life Science & Technology Company, Missouri, TX).
Evaluation of brain edema
Brain edema was evaluated using the wet weight-dry weight measurements of the brain (Toklu et al., 2009b). The whole brain was weighed and then dried for 48 h at 100°C, and then re-weighed. The percentage of water was calculated according to the following formula:% H2O = [(wet weight – dry weight)/wet weight] × 100.
Evans blue assay for the evaluation of blood–brain barrier permeability
To evaluate blood–brain barrier (BBB) integrity, Evans blue dye (EB) was used as a marker of albumin extravasation (Toklu et al., 2009a, 2009b). At 48 h after SAH induction, under ketamine anesthesia, EB (in 2% in saline, 4 mL/kg) was injected via the jugular vein and was allowed to remain in the circulation for 30 min. Then the chests were opened and the rats were perfused transcardially with 250 mL of saline at a pressure of 110 mm Hg for approximately 15 min. After decapitation, the brain was removed and dissected into cerebral cortex and cerebellum, which were then weighed separately for the quantitative measurement of EB-albumin extravasation. Brain samples were homogenized in 2.5 ml phosphate-buffered saline and mixed by vortexing for 2 min after the addition of 2.5 mL of 60% trichloroacetic acid to precipitate the protein. The samples were cooled and then centrifuged for 30 min at 1000g. The supernatant was measured at 620 nm for the absorbance of EB using a spectrophotometer (Shimadzu UV-1208; Shimadzu, Kyoto, Japan). EB was expressed as micrograms per milligram of brain tissue against a standard curve.
Measurement of Na+-K+-ATPase activity
Since Na+-K+-ATPase is a membrane-bound enzyme required for cellular transport, reductions in its activity indirectly indicate membrane damage and impaired cellular function. Measurement of Na+-K+-ATPase activity is based on the measurement of inorganic phosphate released by ATP hydrolysis during incubation of brain homogenates in an appropriate medium containing 3 mM ATP as a substrate. The total ATPase activity was determined in the presence of 100 mM NaCl, 5 mM KCl, 6 mM MgCl2, 0.1 mM EDTA, and 30 mM Tris HCl (pH 7.4), while the Mg2+-ATPase activity was determined in the presence of 1 mM ouabain. The difference between the total and the Mg2+-ATPase activities was taken as a measure of the Na+-K+-ATPase activity (Reading and Isbir, 1980). The reaction was initiated with the addition of the homogenate (0.1 mL), followed by a 5-min pre-incubation period at 37°C. Following the addition of Na2-ATP and a 10-min re-incubation period, the reaction was terminated by the addition of ice-cold 6% perchloric acid. The mixture was then centrifuged at 3500g, and Pi in the supernatant fraction was determined by the method of Fiske and Subbarow (1925). The specific activity of the enzyme was expressed as nmol Pi mg−1 protein h−1. The protein concentration of the supernatant was measured by the Lowry method (Lowry et al., 1951).
Measurement of myeloperoxidase activity
Tissue-associated myeloperoxidase (MPO) activity was determined as an indication of the accumulation of neutrophils. MPO is a natural constituent of primary granules of neutrophils, and a direct relationship between the MPO activity measured in tissue samples and the number of neutrophils was previously shown (Bradley et al., 1982). Brain tissue samples were homogenized in 50 mM potassium phosphate buffer at a pH of 6.0, and centrifuged at 41,400g for 10 min. The pellets were then suspended in 50 mM PB containing 0.5% hexadecyltrimethylammonium bromide. After three freeze-thaw cycles, with sonication between the cycles, the samples were centrifuged at 41,400g for 10 min. Aliquots (0.3 mL) were added to 2.3 mL of reaction mixture containing 50 mM PB, o-dianisidine, and 20 mM H2O2 solution. One unit of enzyme activity was defined as the amount of MPO present that caused a change in absorbance, as measured at 460 nm for 3 min. MPO activity was expressed as units per gram of tissue (Hillegass et al., 1990).
Chemiluminescence (CL) assay
To assess the role of reactive oxygen species (ROS) in SAH-induced brain damage, luminol and lucigenin chemiluminescences were measured as indicators of radical formation. Lucigenin (bis-N-methylacridinium nitrate) and luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) were obtained from Sigma-Aldrich (St. Louis, MO). Measurements were made at room temperature using a Junior LB 9509 luminometer (EG&G Berthold, Bad Wildbad, Germany). Tissue samples cut into small pieces were put into vials containing PBS-HEPES buffer (0.5 M PBS containing 20 mM HEPES, pH 7.2). ROS were quantitated after the addition of the enhancers lucigenin or luminol, for a final concentration of 0.2 mM. Luminol detects a group of reactive species, such as •OH, H2O2, and HOCl radicals, while lucigenin is selective for O2– (Davies et al., 1992). Counts were obtained at 1-min intervals, and the results were given as the area under the curve (AUC) for a counting period of 5 min. Counts were corrected for wet tissue weight and expressed as relative light units (rlu) per milligram of tissue (Ohara et al., 1993).
DNA fragmentation assay
The percentage of DNA fragmentation in the brain tissue was determined as an indicator of cell death, including apoptosis. Brain tissue samples were homogenized in 10 volumes of a lysis buffer (5 mM Tris HCL, 20 mM ethylenediaminetetraacetic acid [EDTA], 0.5% [v/v] t-octylphenoxypolyethoxyethanol [Triton-X 100]; pH 8.0). Two separate samples of 1 mL each were taken from the samples and centrifuged at 25,000g for 30 min to separate the intact chromatin in the pellet from the fragmented DNA in the supernatant (Wyllie, 1980). The supernatant was taken out to be saved and the pellet was re-suspended in 1 mL of Tri-EDTA buffer (pH 8.0), 10 mM:1 mM, respectively. Both the supernatant and the re-suspended pellet were assayed for DNA content by diphenylamine reaction as previously described by Burton (1956).
Malondialdeyde and glutathione assays
Brain samples were homogenized with ice-cold 150 mM KCl for the determination of malondialdehyde (MDA) and glutathione (GSH) levels, indicating lipid peroxidation and intracellular antioxidant status, respectively. The MDA levels were assayed for the products of lipid peroxidation, and the results are expressed as nanomoles MDA per gram of tissue (Beuge and Aust, 1978). GSH was determined by a spectrophotometric method based on the use of Ellman's reagent, and the results are expressed as micromoles GSH per gram of tissue (Beutler et al., 1963).
Histopathological preparation and analysis
Anesthetized (IP ketamine and chlorpromazine) rats were perfused transcardially with a solution of 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4). To obtain basilar artery sections, the anterior midline of the brainstem was removed. For light microscopic analysis, perfused brain specimens were fixed in 10% formaldehyde, dehydrated in an alcohol series, cleared in toluene, and embedded in paraffin. Paraffin sections (5 μm) were stained with hematoxylin and eosin, and examined under a photomicroscope (Olympus BH 2; Olympus, Tokyo, Japan).
For the electron microscopic evaluation, tissues obtained from perfused rats were post-fixed with 1% OsO4, dehydrated in an alcohol series, and embedded in Epon-812 resin. Thin (1 μm) sections stained with toluidine blue, and 60-nm sections stained with 1% lead citrate and uranyl acetate, were examined under a JEOL 5200 JSM (JEOL Ltd., Tokyo, Japan) electron microscope.
Statistical analysis
Statistical analysis was done using GraphPad Prism 3.0 (GraphPad Software, San Diego, CA). All data are expressed as means ± SEM. Groups of data were compared with an analysis of variance (ANOVA), followed by Tukey's multiple comparison test. Values of p < 0.05 were considered statistically significant.
Results
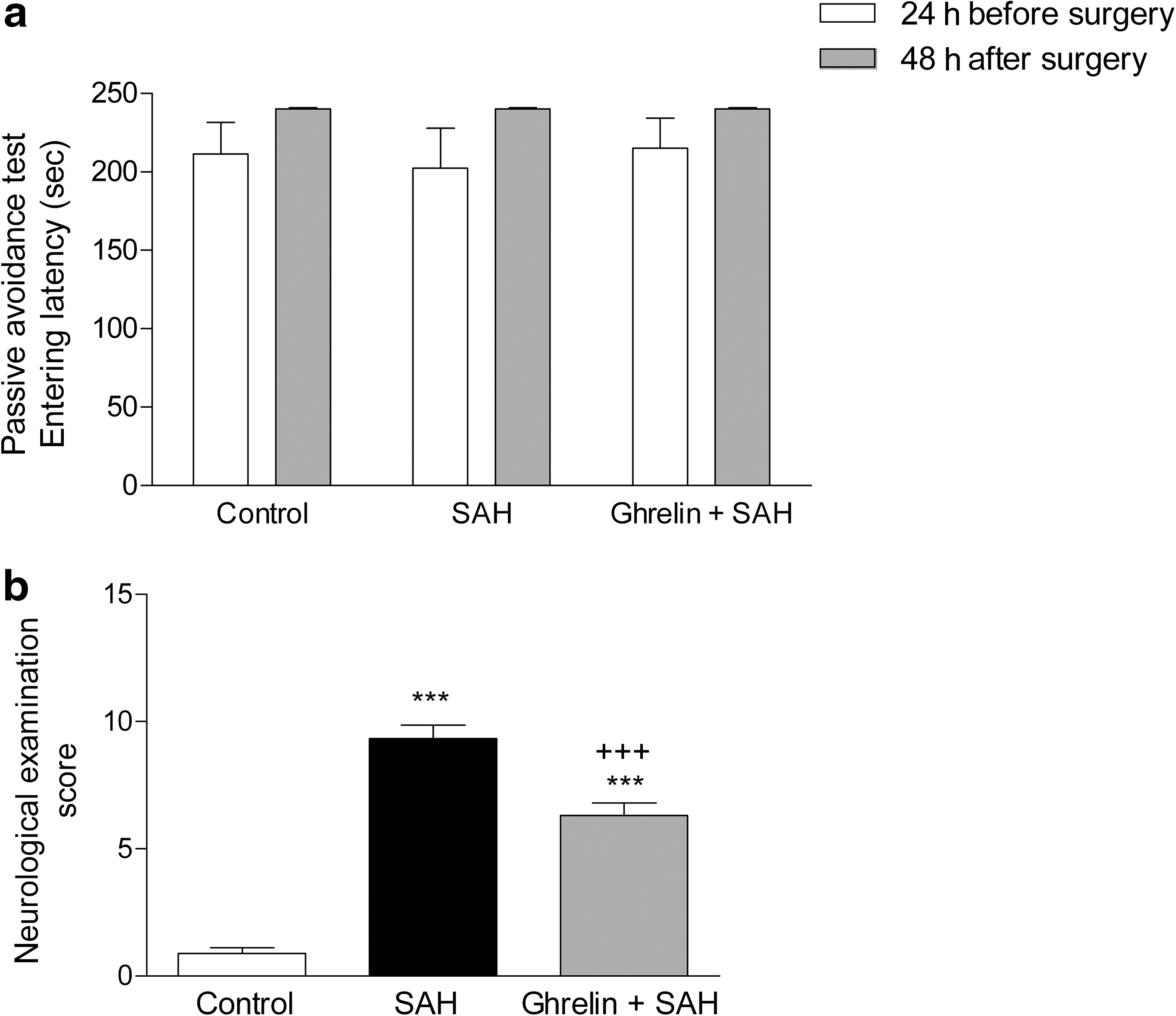
Passive avoidance testing results revealed that memory function of the rats that underwent SAH surgery were not altered, because the rats in all groups did not enter the dark compartment until the cut-off time of the second trial performed at the 48 h post-surgery (Fig. 1a). However, the average neurological examination scores recorded at 48 h post-surgery was significantly higher in the saline-treated SAH group than in the sham-operated control group (p < 0.001). On the other hand, the average score was significantly reduced in the ghrelin-treated SAH group (p < 0.001; Fig. 1b), but was still higher than that of the sham-operated control group.
(
In the saline-treated SAH group, TNF-α and IL-1β levels measured 3 and 48 h after SAH were significantly increased compared to controls (p < 0.01–0.001), while these SAH-induced elevations in plasma proinflammatory cytokines were reduced (p < 0.05–0.001), when the rats received ghrelin treatment after SAH induction (Fig. 2). Plasma levels of S-100β protein and NSE measured 3 h after SAH induction were significantly elevated in the saline-treated group (p < 0.001; Fig. 3), while these elevations were suppressed in the ghrelin-treated SAH group (p < 0.01). At 48 h after SAH-induced brain injury, the plasma NSE level in the saline-treated group was found to be still elevated compared to the control group (p < 0.001), while the plasma NSE level in the ghrelin-treated SAH group was no different than that of the control group. On the other hand, plasma S-100β was reduced back to control levels in both the saline- and ghrelin-treated SAH groups.
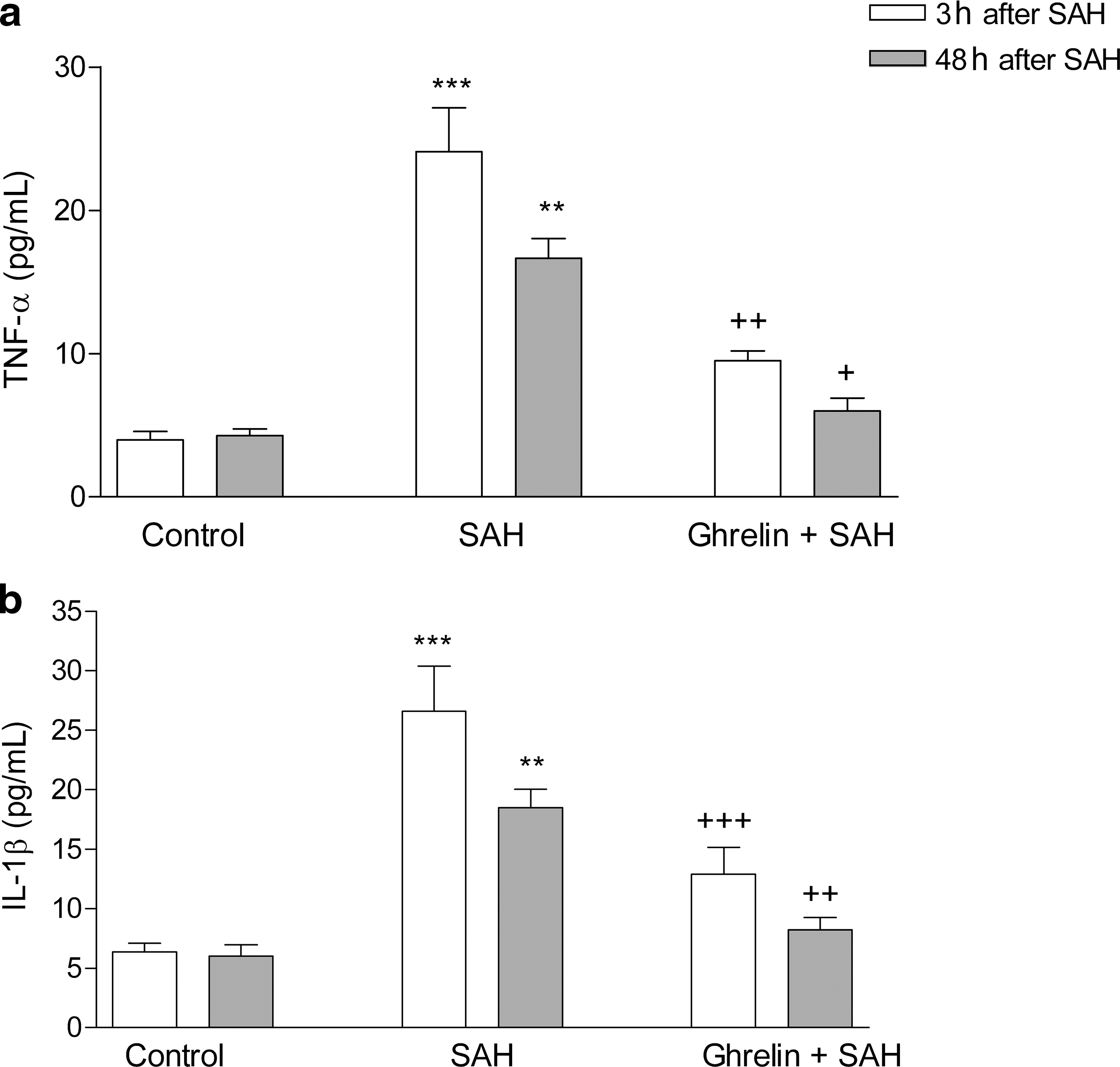
Plasma (
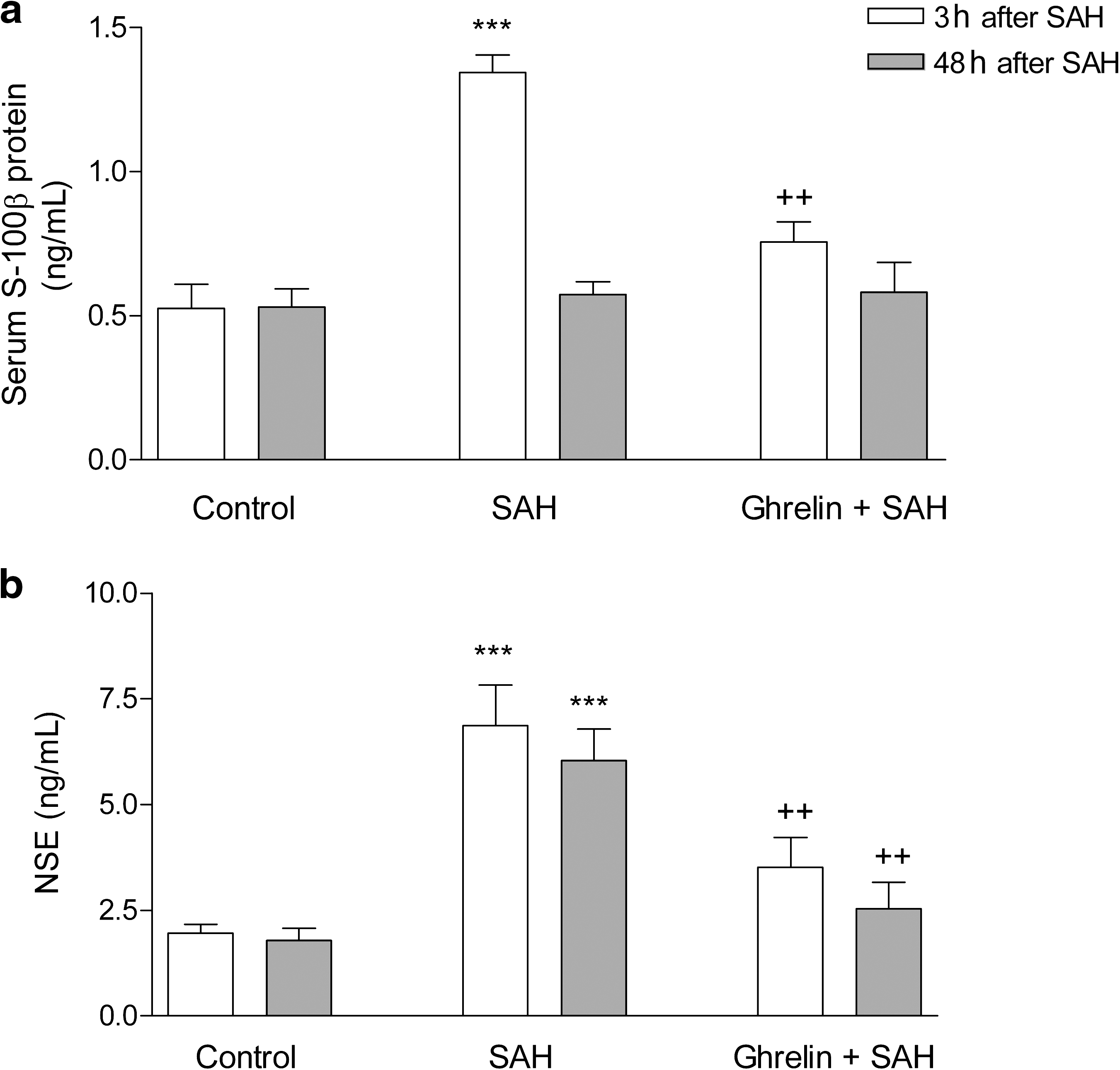
Plasma (
Brain water content and EB content were higher in the saline-treated SAH group than in the control group (p < 0.001), while SAH-induced brain edema and increased BBB permeability were reduced back to control levels in the ghrelin-treated group (p < 0.05; Fig. 4). In addition, in the SAH group, Na+-K+-ATPase activity was significantly depressed (p < 0.01), implicating an impaired membrane transport function, while the activity of the pump in the ghrelin-treated SAH group was no different than that of the control rats (Fig. 4c).

(
Myeloperoxidase activity in the cerebral tissue was significantly elevated in the saline-treated SAH group (p < 0.001; Fig. 5a), while in the ghrelin-treated group it had reverted to the activity level of the control group (p < 0.05). Similarly, SAH induction caused significant elevations in the brain luminol and lucigenin CL levels (p < 0.01), indicating enhanced generation of ROS in the cerebral tissue, but these increases were significantly suppressed by ghrelin treatment (p < 0.01; Fig. 5b and c).

(
In the cerebral tissue samples of the saline-treated SAH group, DNA fragmentation was significantly increased compared to the control group (p < 0.001; Fig. 6a), while the SAH-induced increase in DNA fragmentation was suppressed by ghrelin treatment (p < 0.05). Furthermore, in the SAH-induced group that received saline, the brain MDA level was markedly increased, indicating enhanced lipid peroxidation compared to sham-operated controls (p < 0.001; Fig. 6b), while ghrelin administration reduced this SAH-induced elevation (p < 0.05). In accordance with this finding, SAH caused a significant depletion of brain GSH content compared to controls (p < 0.01; Fig. 6c), but the brain GSH content in the ghrelin-treated SAH group was partially preserved.

(
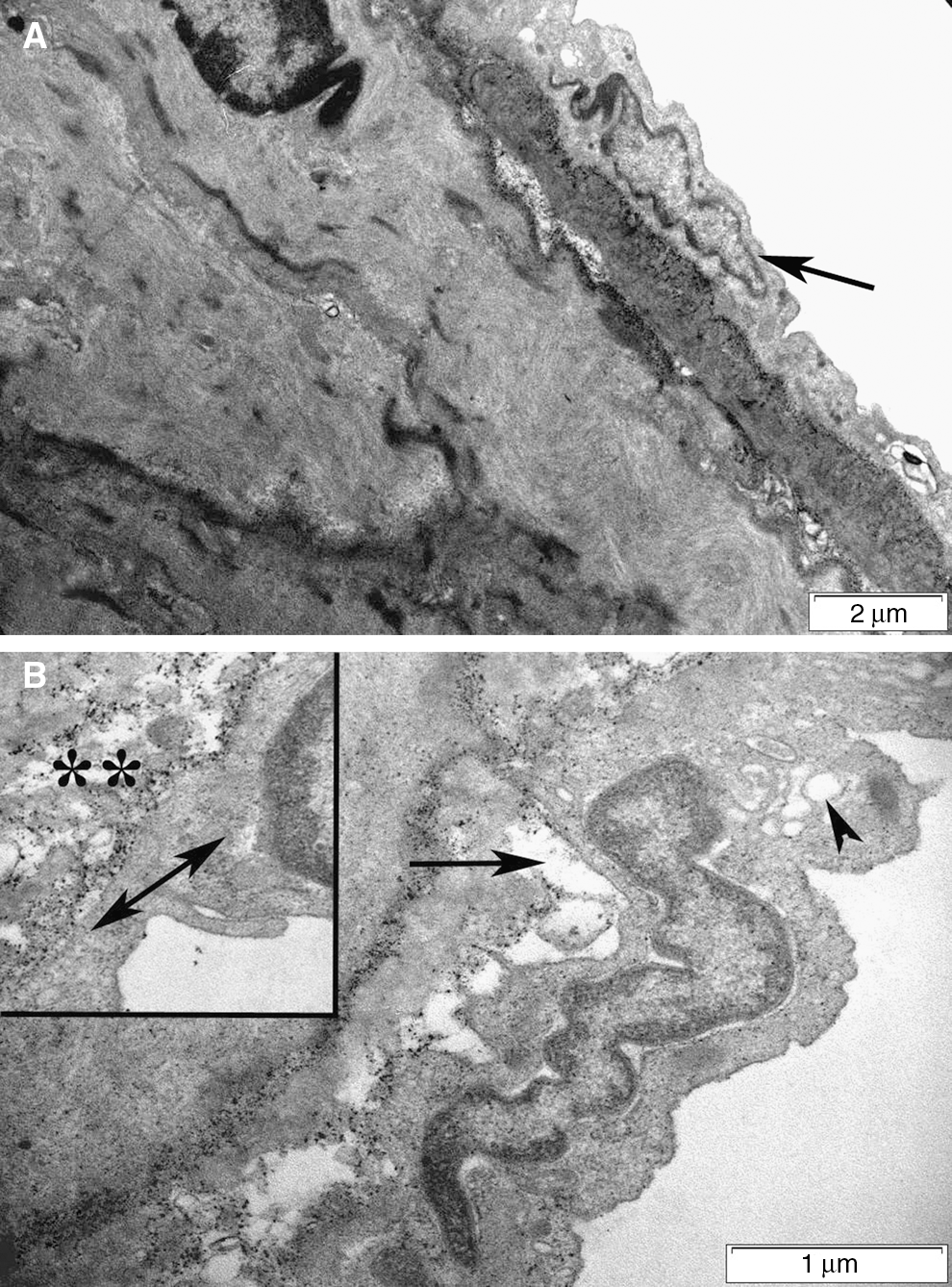
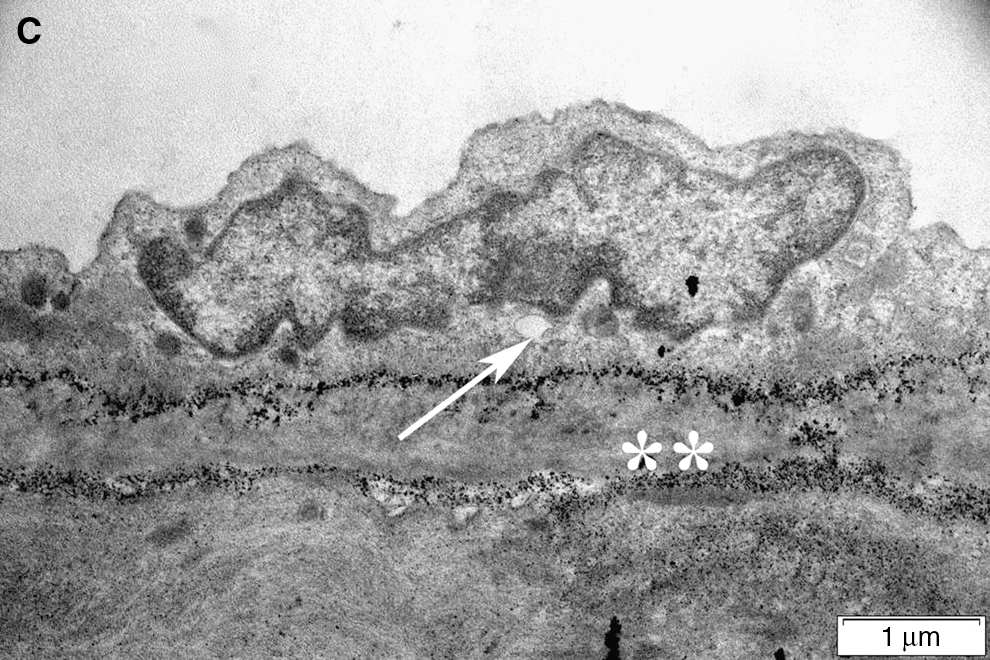
Light microscopic evaluation revealed regular alignment of the basilar arteries with endothelial cells and elastic lamina in the control group (Fig. 7a), while in the SAH group, vasospasm of the arteries was clear, and the continuity of both the endothelium and elastic lamina showed focal disruptions indicating degeneration (Fig. 7b). On the other hand, decreased arterial vasospasm with improved integrity of the endothelium was prominent in the ghrelin-treated group (Fig. 7c). Accordingly, transmission electron microscopic examination of SAH injury demonstrated severe degeneration in the endothelium, elastic lamina, and connective tissue of the basilar artery (Fig. 8b), compared to controls (Fig. 8a), with intense edema and vacuolization in the cytoplasm of both endothelial and muscle cells. Detachments between the endothelial cytoplasm and the elastic lamina were prominent. The nuclei of both endothelial and muscle cells showed contractions due to spasm of the artery. Ghrelin treatment ameliorated the injury, and the contraction of the artery was reduced, with a concomitant decrease in cytoplasmic vacuolization and edema (Fig. 8c).
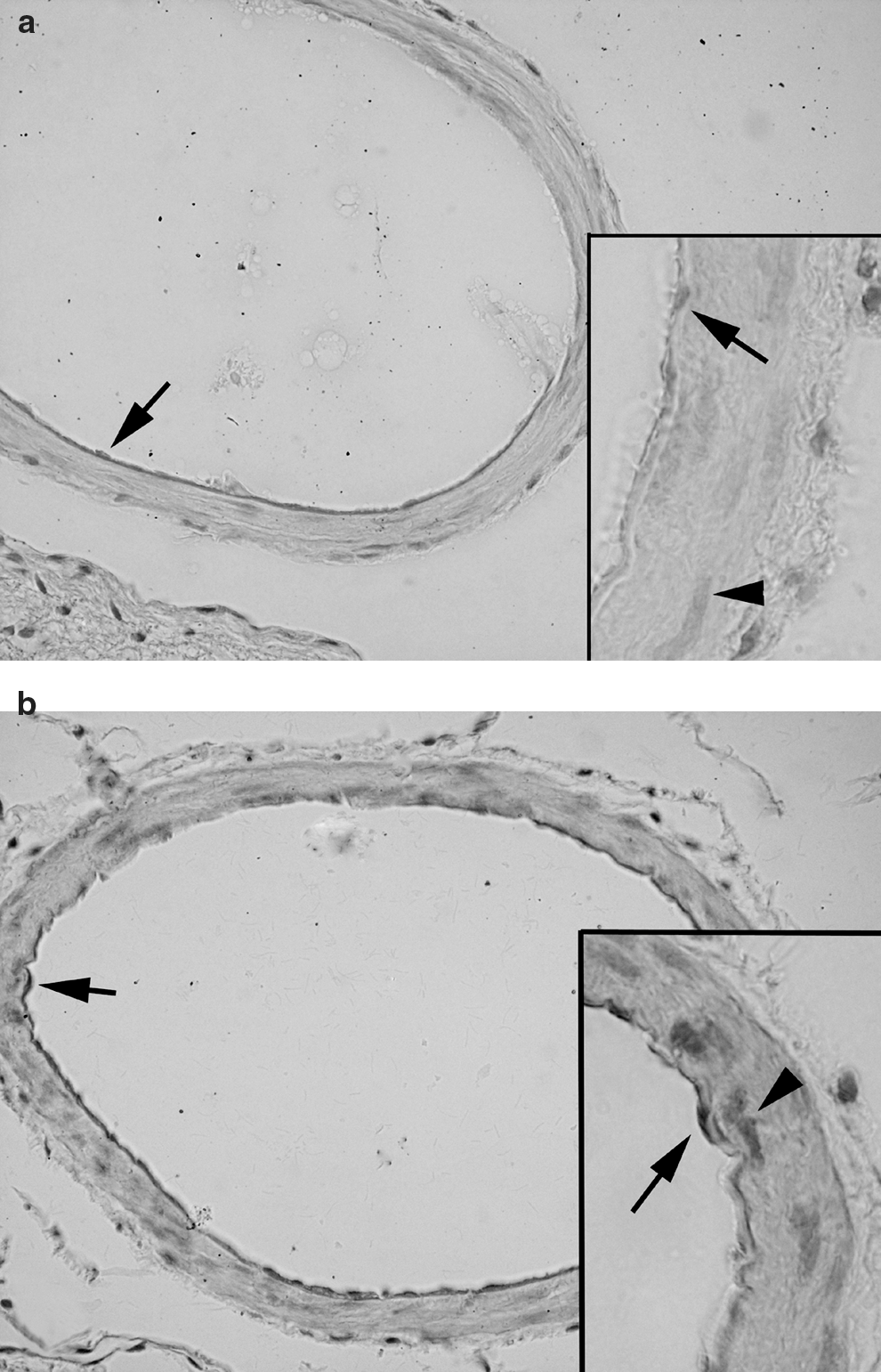
Light microscopy. (

Transmission electron microscopy. (
Discussion
The results of the current study demonstrate that SAH-induced neurological impairment and oxidative brain injury, as evidenced by increased generation of ROS, increased tissue MDA levels, DNA fragmentation, and MPO activity, along with reduced GSH content and Na+-K+-ATPase activity, were ameliorated by ghrelin treatment. Furthermore, SAH-induced morphological changes in the basilar arteries were improved by ghrelin treatment, while SAH-induced elevations in the plasma levels of TNF-α and IL-1β, as well as NSE and S-100β protein, were also reduced by ghrelin treatment. These findings suggest that ghrelin, by inhibiting MPO activity and the subsequent release of inflammatory mediators that induce lipid peroxidation, appears to play a neuroprotective role in SAH-induced injury in rats.
Ghrelin, secreted predominantly by the mucosal endocrine cells of the stomach, and identified as the endogenous ligand of the growth hormone secretagogue receptor (GHS-R), has profound orexigenic, adipogenic, and somatotropic properties (Kojima et al.,1999; Ukkola, 2009). The wide distribution of ghrelin in various organs, including lymphoid tissues (Date et al., 2000; Hattori et al., 2001), suggests that it has other functions not directly associated with appetite control. Ghrelin has been shown to exhibit anti-inflammatory functions in vitro (Chorny et al., 2008; Dixit et al., 2004; Wassem et al., 2008), and in several animal models by enhancing immune responses and downregulating anti-inflammatory molecules (Chorny et al., 2008; Gonzalez-rey et al., 2006; Granado et al., 2005; Li et al., 2004). Administration of a GHS-R agonist in a mouse model of arthritis resulted in a decrease in IL-6 levels and reduced signs of inflammation (Granado et al., 2005). Since ghrelin receptors have been found in peripheral tissues such as bone marrow, spleen, and lymphocytes (Gnanapavan et al., 2002; McKee et al., 1997), the anti-inflammatory effects of the GHS-R agonist were suggested to be mediated by ghrelin receptors directly expressed in T lymphocytes and monocytes, where ghrelin inhibits the expression of proinflammatory cytokines (Dixit et al., 2004). In support of these reports, our findings demonstrate that ghrelin reduces plasma levels of proinflammatory TNF-α and IL-1β in the setting of SAH-induced inflammation.
Despite sophisticated neurosurgical management of ruptured intracranial aneurysms, SAH is associated with high mortality and severe morbidity rates, and causes severe cognitive and psychosocial impairments (Hütter et al., 2001). Aneurysmal SAH frequently results in complications, including intracranial hypertension, rebleeding, and vasospasm. The extravasated blood is responsible for a cascade of reactions involving the release of various vasoactive and proinflammatory factors from blood and vascular components in the subarachnoid space (Sercombe et al., 2002). Following the global ischemia seen with SAH, apoptosis has been shown to occur in the hippocampus, BBB, and vasculature, with varying degrees of necrosis (Park et al., 2004). Breakdown of the BBB, which is known to disrupt the microenvironment of the central nervous system (CNS), has been detected in many neurodegenerative diseases and traumatic brain injuries, in which the release of ROS and proinflammatory cytokines play an important role (Belayev et al., 1996; Huber et al., 2001; Panikashvili et al., 2006; Whitton, 2007). Belayev and colleagues (1996) postulated that free radicals lead to an increase in BBB permeability by attacking the polyunsaturated fatty acids of the endothelial cell membrane, which then results in vasogenic edema and hemorrhage. In the present study, SAH resulted in neurological impairment accompanied by brain edema and increased permeability of the BBB, while enhanced chemiluminescence results showed that SAH-induced injury facilitated free-radical generation, with a concomitant increase in brain MDA levels. Moreover, MPO activity, utilized as an indirect index of neutrophil infiltration, was increased at 48 h post-SAH, suggesting the involvement of neutrophils in SAH-induced oxidative brain injury. Accordingly, it has been demonstrated that increased BBB permeability due to SAH results in the accumulation of blood cells such as neutrophils and macrophages in the brain (Erşahin et al., 2009), which further sustains the cerebral inflammatory cascade. The neutrophils activate the NADPH oxidase and MPO enzyme systems, causing production of large amounts of HOCl, which oxidizes and damages macromolecules, including proteins, lipids, carbohydrates, and nucleic acids (Dietrich and Dacey, 2000; Reiter et al., 2001). On the other hand, the current findings demonstrate that ghrelin treatment depressed MPO activity and ROS generation, and helped to repair the BBB, suggesting that the protective effects of ghrelin in SAH-induced brain injury is mediated in part by blocking tissue neutrophil infiltration and the associated ROS-induced damage. In accordance with the present data, it was previously shown that ghrelin ameliorates organ injury in several inflammation models by inhibiting proinflammatory cytokine production and myeloperoxidase activity (Brzozowski et al., 2006; Iseri et al., 2008; Kasımay et al., 2006; Sehirli et al., 2008; Shah et al., 2009; Wu et al., 2008). Furthermore, ghrelin has been shown to exhibit anti-inflammatory functions against T cells and macrophages by inhibiting mononuclear cell binding and nuclear factor-κB activation (Chorny et al., 2008; Dixit et al., 2004; Li et al., 2004; Wassem et al., 2008). Recently, treatment with ghrelin was shown to suppress mRNA levels of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 in microglia and infiltrating T cells derived from spinal cord in an experimental model of autoimmune encephalitis (Theil et al., 2009), suggesting an immunomodulatory role for ghrelin in CNS pathologies that involve the production of proinflammatory cytokines.
Under physiological conditions, cellular defense systems, such as GSH and GSH-related antioxidant enzymes, protect brain tissue from the damaging effects of ROS, while decreased levels of GSH have been implicated in the pathogenesis of SAH-induced vasospasm (Kamezaki et al., 2002; Marzatico et al., 1990; Pyne-Geithman et al., 2009; Sano et al., 1980). Accordingly, SAH-induced oxidative injury in the current study was associated with a reduction in brain GSH content. On the other hand, the findings revealed that the improved neurological state and attenuated oxidative brain injury in ghrelin-treated rats was accompanied by the maintenance of brain GSH stores. Previously, ghrelin pretreatment has been reported to diminish oxidative stress and prevent the decrease in antioxidant enzyme activity seen in a rat seizure model (Obay et al., 2008). Similarly, ghrelin was shown to preserve brain GSH content in other models of oxidative brain damage (Liu et al., 2006; Miao et al., 2007; Xu et al., 2009).
SAH is a complex phenomenon with a prolonged course, in which the early ischemic response is followed by a secondary wave of cell death due to the neuroinflammatory response to the initial injury. Serum levels of NSE and S-100β are early predictors of outcome in head-injured and stroke patients (Beaudeux, 2009; Oertel et al., 2006; Raabe et al., 1999; Stroick et al., 2006; Townend and Ingebrigtsen, 2006), and their elevation reflects increased severity of the primary hypoxic-ischemic insult (Hatfield and McKernan, 1992; Hardemark et al., 1988, 1989; Kaiser et al., 1989; Mayer and Linares, 2009). The neuronal marker studied most extensively in cerebral ischemia is NSE, a neuron-specific intracytoplasmic enzyme in the glycolytic pathway, which is released into cerebrospinal fluid (CSF) and blood after cerebral injury (Cunningham et al., 1991; Horn et al., 1995; Marangos et al., 1976). Serum and CSF NSE levels were shown to be elevated in rodent models of focal ischemia in proportion to the eventual infarct volume (Barone et al., 1993; Steinberg et al., 1984). Moreover, increases in the peripheral levels of NSE have been specifically related to neuronal injury in traumatic brain injury (Herrmann et al., 1999.), stroke (Persson et al., 1987), and epileptic seizures (Rabinowicz et al., 1996). Another glycolytic pathway enzyme, S100-B, is released mainly from astrocytes in multiple forms of CNS damage, including ischemic stroke, CNS trauma, and neurodegenerative diseases (Rothermundt et al., 2003). In the current study, serum S100-B and NSE levels were shown to be elevated at 2 and 48 h after SAH-induced injury. On the other hand, ghrelin treatment reduced plasma levels of these neuronal injury indicators and suppressed neuronal death as assessed by the DNA fragmentation ratio. Moreover, the significant decrease in Na+-K+-ATPase activity, indicating membrane damage and deterioration of membrane fluidity, was also reversed by ghrelin treatment. Inhibition of this crucial enzyme activity is found in several neuropathological conditions, including oxidative brain injury (Erşahin et al., 2009; Franzon et al., 2003), and cerebral vasospasm (Erşahin et al., 2009). Since Na+-K+-ATPase at high concentrations in the brain are responsible for the generation of the membrane potential through the active transport of sodium and potassium ions (Brillault, 2008; Erecinska et al., 2004; Foroutan, 2005), the inhibition of this pump under ischemic conditions, possibly by free-radical attack (Kurella et al., 1997; Yu et al., 2003), promotes the reversal of the neurotransmitter transporters, contributing to the rise in the extracellular concentration of different transmitters seen during brain ischemia (Foroutan, 2005; Santos et al., 1996). However, despite the depression in Na+-K+-ATPase activity and the subsequent neuronal injury, SAH-induced oxidative damage did not alter memory function in the current study. Similarly, only a slight cognitive impairment was seen in rats that survived for 5 weeks following SAH (Takata et al., 2008). Since memory function was intact 48 h after SAH-induced cerebral injury, the previously-demonstrated stimulatory effects of centrally-administered ghrelin on memory retention (Carlini et al., 2008; Tóth et al., 2009) were not seen. Thus, the influence of ghrelin on learning processes and memory needs to be further explored in chronic SAH.
In conclusion, our results support the anti-inflammatory and neuroprotective effects of ghrelin in SAH-induced oxidative damage, by maintaining a balance in oxidant-antioxidant status through the augmentation of endogenous antioxidants and the inhibition of proinflammatory mediators. Since targeting neuroinflammatory mechanisms of SAH appears to be a promising avenue for therapeutic interventions, ghrelin, which can cross all morphophysiological barriers including the BBB, and has low toxicity and high efficacy (Cunningham et al., 1991; Liu et al., 2006; Miao et al., 2007; Obay et al., 2008; Xu et al., 2009), appears to be a promising new therapeutic strategy for the treatment of SAH-induced oxidative brain injury. Further elucidation of the functions of ghrelin will likely offer new approaches to the treatment of similar neuroinflammatory events.
Author Disclosure Statement
No conflicting financial interests exist.