
Abstract
Traumatic brain injury (TBI) causes a wide spectrum of consequences, such as microglial activation, cerebral inflammation, and focal and diffuse brain injury, as well as functional impairment. In this study we aimed to investigate the effects of acute treatment with minocycline as an inhibitor of microglial activation on cerebral focal and diffuse lesions, and on the spontaneous locomotor activity following TBI. The weight-drop model was used to induce TBI in mice. Microglial activation and diffuse axonal injury (DAI) were detected by immunohistochemistry using CD11b and ß-amyloid precursor protein (ß-APP) immunolabeling, respectively. Focal injury was determined by the measurement of the brain lesion volume. Horizontal and vertical locomotor activities were measured for up to 12 weeks post-injury by an automated actimeter. Minocycline or vehicle were administered three times post-insult, at 5 min (90 mg/kg i.p.), 3 h, and 9 h post-TBI (45 mg/kg i.p.). Minocycline treatment attenuated microglial activation by 59% and reduced brain lesion volume by 58%, yet it did not affect DAI at 24 h post-TBI. More interestingly, minocycline significantly decreased TBI-induced locomotor hyperactivity at 48 h post-TBI, and its effect lasted for up to 8 weeks. Taken together, the results indicate that microglial activation appears to play an important role in the development of TBI-induced focal injury and the subsequent locomotor hyperactivity, and its short-term inhibition provides long-lasting functional recovery after TBI. These findings emphasize the fact that minocycline could be a promising new therapeutic strategy for head-injured patients.
Introduction
T
Sequelae of TBI include a wide spectrum of effects on the injured brain. TBI causes focal and diffuse brain injury, cerebral inflammation, and hemorrhage, all of which may lead to acute and chronic cell death, and may contribute to functional impairment (Bye et al., 2007; Hellal et al., 2003; Homsi et al., 2009; Werner and Engelhard, 2007; Zhang et al., 2005). Diffuse axonal injury (DAI) constitutes one of the main consequences of TBI. It is thought to account for most of the neurological disability and loss of consciousness seen following TBI (Li and Feng, 2009; Meythaler et al., 2001). DAI typically occurs when the head is subjected to shear-strain forces, with most lesions emerging at the interface between regions of the brain that have different tissue densities, such as at the junctions between grey and white matter (Li and Feng, 2009). The shearing forces associated with closed head injury induce axonal alterations, which may extend to axonal swelling and disruption, detachment of distal axons, and formation of axonal retraction bulbs (Büki and Povlishock, 2006; Farkas and Povlishock, 2007; Medana and Esiri, 2003). Hypoxia, ischemic brain damage, cerebral edema, and cerebral herniation are often secondary complications seen following head injury, and all may play a role in the progression of axonal injury. DAI can be detected by the axonal accumulation of ß-amyloid precursor protein (ß-APP) within hours following TBI in rodents (Stone et al., 2000), and in post-mortem brain samples of head-injured patients (Gentleman et al., 1993; Sherriff et al., 1994). Therefore, axonal ß-APP immunostaining is considered to be the best marker of DAI (Gentleman et al., 1993; Sherriff et al. 1994; Stone et al., 2000). ß-APP is a ubiquitously expressed transmembrane glycoprotein synthesized within the neuronal cytoplasm by the Golgi apparatus (Selkoe et al., 1994). It is carried along axons to the synapse by a fast anterograde axoplasmic transport, and upon injury it rapidly pools at the site of focally-impaired tissue, and accumulates in axonal bulbs (Medana and Esiri, 2003).
Simultaneously, TBI almost immediately triggers an immune response, that includes the activation of resident glial cells, microglia, and astrocytes, which secrete soluble inflammatory cytokines (Morganti-Kossmann et al., 2007). Notably, the TBI-induced inflammatory response (i.e., the rapid release of TNF-α from activated microglia) may contribute to early axonal accumulation of ß-APP (Kita et al., 2000). Therefore inhibition of the inflammatory response by tacrolimus (Thomale et al., 2007; Zawadzka and Kaminska, 2005) may lead to a reduction in axonal injury following TBI (Marmarou and Povlishock, 2006; Singleton et al., 2001), suggesting that neuroinflammation may play a crucial role in the progression of DAI following TBI.
Cerebral inflammation plays a dual opposing role, by supporting the processes of repair on one hand, and by exacerbating tissue damage on the other (Morganti-Kossmann et al., 2007). Microglia constitute the macrophage equivalent of the central nervous system (CNS) (Kreutzberg, 1996; Streit, 2000), and are estimated to account for 5–20% of the total glial population, but they are less numerous in the white matter than in the grey matter (Lynch, 2009; van Rossum and Hanisch, 2004). Microglia derive from mesodermal precursor cells of the hematopoietic lineage that populates the CNS in early development, and therefore they share the phenotypic markers of monocytes and macrophages (Liu and Hong, 2003; Lynch, 2009). In the mature brain and under physiological conditions, resting microglia adopt their characteristic ramified appearance and play a role in immune surveillance and host defense. However, since microglia are particularly sensitive to changes in their microenvironment, they readily become activated and adopt an amoeboid macrophage-like morphology in response to brain infection or injury, such as cerebral ischemia or TBI (Hansson and Rönnbäck, 2003; Napoli and Neumann, 2009). Microglia secrete anti-inflammatory and proinflammatory mediators, and generate reactive oxygen species and nitrogen intermediates. Therefore intense microglial activity is harmful, since it can induce secondary brain damage (Aloisi, 2001; van Rossum and Hanisch, 2004). It is known that multiple pathways such as oxidative stress, nitrosative stress, neuroinflammation, and apoptosis may be triggered upon microglial activation, and may lead to detrimental consequences following brain injury, such as the amplification of axonal injury (Kelley et al., 2007; Medana and Esiri, 2003; Zhang et al., 2005). Therefore, the short-term prevention of excessive microglial activation during the acute phase of TBI may prove to be beneficial. In fact, the timing and the length of the inhibition of microglial activation may affect the risk:benefit ratio and the final TBI outcome.
One of the well-known compounds used during the last decade to suppress microglial activation is minocycline, a highly lipophilic second-generation derivative of the antibiotic tetracycline (Kim and Suh, 2009). It is thought that minocycline exerts anti-inflammatory and neuroprotective effects in a number of animal models of brain injury and neurodegenerative disease (Domercq and Matute, 2004; Elewa et al., 2006; Kim and Suh, 2009; Yong et al., 2004), despite some contradictory or even detrimental effects that have been reported in recent years (Diguet et al., 2004; Gordon et al., 2007). The proposed mechanisms of minocycline's neuroprotective action include the attenuation of microglial activation (Tikka and Koistinaho, 2001; Yrjanheikki et al., 1998, 1999), the inhibition of the activation of the p38 mitogen-activated protein kinase (Tikka and Koistinaho, 2001), and the subsequent decrease in cytokine, chemokine, matrix metalloproteinase (MMP), and nitric oxide production, inhibition of MMP-9 activity (Romero-Perez et al., 2008), potent inhibition of poly(ADP-ribose) polymerase-1 activity (Alano et al., 2006), depression of glutamatergic calcium signaling (González et al., 2007; Tikka and Koistinaho, 2001), antioxidant activity (Kraus et al., 2005), anti-apoptotic effects of the inhibition of the expression of caspase-1 and caspase-3 (Chen et al., 2000), and inhibition of cytochrome c release from mitochondria via the blockade of the opening of mitochondrial permeability transition pores (Zhu et al., 2002).
However, among acute brain injuries, treatment of TBI with minocycline has received little interest in experimental studies. To date, treatment with minocycline has only been shown to reduce brain lesion volume, neurological dysfunction, and caspase-1 activation (Sanchez Mejia et al., 2001). A transient neuroprotection induced by minocycline has also been reported, in association with a reduction in microglial activation and interleukin-1ß production (Bye et al., 2007). Recently, we showed that acute treatment with minocycline was able to reduce cerebral edema, and that it reduced production of certain inflammatory markers (Homsi et al., 2009). However, no study has focused on the effects of minocycline on TBI-induced DAI, and on the long-term functional outcome following TBI.
Therefore in the present study we evaluated the time course of microglial activation and DAI following an experimental model of closed-head injury. Then we investigated the effects of acute treatment with minocycline on microglial activation, on DAI, and on focal brain lesions at 24 h post-TBI. Additionally, the long-term effects of minocycline on post-TBI functional outcome was assessed for up to 12 weeks post-injury. Our findings highlighted the beneficial effects of the acute blockade of microglial activation by minocycline on focal lesion volume, although without affecting DAI, and more interestingly its effect on functional outcome, by reducing post-TBI locomotor hyperactivity over the long term.
Methods
Animals
All experiments were performed on male Swiss mice (Janvier, Le Genet St. Isle, France) weighing 28–30 g, that were housed in a temperature-controlled environment (22 ± 2°C), with a 12-h light/dark cycle, and access to food and water ad libitum. Animal care and all experiments were carried out with the ethical approval of the University of Paris Descartes Animal Ethics Committee, and in accordance with the French regulations and the European Communities Council Directive of November 24, 1986 (86/609/EEC) for the protection of animals for experimental use.
Closed-head injury model
The mouse model of closed-head injury was used as previously described (Hall, 1985; Hellal et al., 2003, 2004; Homsi et al., 2009). The mice subjected to TBI were briefly anesthetized with halothane (2%) and closed-head trauma was induced by a 50-g weight dropped from a height of 36 cm along a stainless steel track impacting the right frontal side of the head. This model is generally associated with a 5–15% mortality rate within the first 5 min following impact, and no delayed mortality was observed thereafter. For the kinetic studies, the animals were randomly divided into six different groups as follows: naive, TBI at 6, 24, 48, and 72 h, and 7 days after injury (n = 5–6 per group and per time point). For the histological analyses, the animals were subjected to cervical dislocation, and the brains were rapidly removed and placed on ice as previously described (Hellal et al., 2003), and frozen in isopentane and stored at −40°C until use.
Immunohistochemistry of microglial activation and diffuse axonal injury
The brains were cut into 20-μm-thick slices using a cryostat at −20°C. Several coronal brain slices were collected at 0.3-mm intervals from A6.26 to A2.96 mm anterior to the interaural line. For our immunohistochemistry studies, the slides were fixed in ice-chilled acetone for 5 min, and they were kept at −20°C until use.
CD11b is used as a cell-surface marker of microglial cells, as it increases following microglial activation (Hansson and Rönnbäck, 2003; Liu and Hong, 2003; Lynch, 2009). ß-amyloid precursor protein (ß-APP) is used as a marker of DAI since its axonal transport is impaired following TBI, leading to an axonal accumulation of ß-APP (Gentleman et al., 1993; Sherriff et al., 1994; Stone et al., 2000).
Monoclonal rat anti-mouse CD11b antibody (1:800, MCA711; Serotec, Raleigh, NC), and polyclonal rabbit anti ß-APP antibody (1:1000, 51–2700; Invitrogen Corp., Carlsbad, CA) were used as primary antibodies. Biotinylated polyclonal goat anti-rat antibody (1:400, STAR80B; Serotec), and biotinylated polyclonal donkey anti-rabbit antibody (1:400, RNP1004; Amersham, Louisville, CO) were used as secondary antibodies for CD11b and ß-APP antibodies, respectively. Primary and secondary antibodies were diluted in a solution of PBS-goat serum 1% and PBS-donkey serum 1% for CD11b and ß-APP antibodies, respectively. Detection of labeled antigens was performed with an avidin-biotin horseradish peroxidase kit (ABC VECTASTAIN®, Vector Laboratories, Burlingame, CA), and diaminobenzidine (DAB; Sigma-Aldrich Co., St. Louis, MO) staining, according to manufacturer instructions. Additionally, some sections were incubated in the absence of primary antibody in order to evaluate the nonspecific immunostaining. In both control paradigms for CD11b and ß-APP, no labeled cells were detected.
Immunolabeled cells were counted either for CD11b, in seven sections of the entire ipsilateral right hemisphere (from A5.66 to A3.86 mm anterior to the interaural line), or for ß-APP, in five or eight sections of a sampling area delineated by a 2.85 mm2 rectangle of the corpus callosum (from A5.06 to A3.86 mm in the kinetic study, and from A5.06 to A2.96 mm in the treatment study), with a light microscope (Axioskop; Zeiss, Gottingen, Germany). The results were expressed as the total number of immunolabeled cells or axons per animal brain, obtained by adding together each section's cell count.
Assessment of brain lesion volume
Brain sections were stained with cresyl violet. The lesion volume was measured using 12 sections, collected from A6.26 to A2.96 mm anterior to the interaural line, at 0.3-mm intervals. Ipsilateral olfactory bulbs, olfactory cortex, and striatum are reproducibly insulted in this model. Briefly, the surface of each section's unstained lesion area was delineated and measured using Image J software (National Institutes of Health, Bethesda, MD). Each lesion area was then multiplied by the ratio of the surface of the contralateral (left) to the ipsilateral (right) hemispheres at the same level to correct the lesion area for brain swelling (Golanov and Reis, 1995). The distances between the respective coronal sections were used to calculate the lesion volume. The brain lesion volume (mm3) was then calculated by linear integration of the edema-corrected lesion areas (Golanov and Reis, 1995; Wahl et al., 1997).
Post-traumatic treatment protocols with minocycline
The animals were randomly divided into three groups as follows: naive, vehicle-treated TBI, and minocycline-treated TBI, and the investigator was blinded to group identity. Minocycline or its vehicle, phosphate-buffered saline (PBS; 0.1 M, pH 7.4), were administered three times post-insult, at 5 min (90 mg/kg i.p.), and at 3 and 9 h post-TBI (45 mg/kg i.p.). The treatment protocol and doses of minocycline used in this study have been previously shown to be anti-edematous and anti-inflammatory in our model of TBI (Homsi et al., 2009). The same set of animals was used to evaluate microglial activation, DAI, and focal brain lesion size at 24 h post-TBI (n = 6–7 per group), the time when microglial activation and DAI reach their maximum in our model. A second set of animals was used to evaluate spontaneous locomotor activity at 2 days, and 1, 2, 4, 6, 8, 10, and 12 weeks post-TBI (n = 10–12 per group).
A third set of animals was used to determine if minocycline had any effect on the spontaneous locomotor activity in naïve mice. Therefore, two groups of naïve mice received either vehicle (PBS 0.1 M, pH 7.4, i.p.), or minocycline (n = 8 per group). Minocycline was administered at a dose of 90 mg/kg i.p., followed by a second dose of 45 mg/kg i.p. 3 h later, and minocycline's effects on spontaneous locomotor activity were measured 1 h after the second dose of minocycline.
Measurement of locomotor activity by an automated actimeter
The mice were individually placed in plastic transparent activity compartments (19 × 11 × 14 cm; Imetronic, Bordeaux, France) that were isolated from noise and put under a light intensity of 5 lux. The displacements were measured by photocell beams located across the long axis, 20 mm above the floor (to detect horizontal activities), and in the side of each compartment (to detect vertical activity). The animals were not pre-habituated to the boxes, and their spontaneous locomotor activities were recorded during 60 min. The results are expressed as the number of photocell beam interruptions per 60 min (counts/60 min) for both horizontal and vertical activities.
Statistical analysis
Data are expressed as mean ± SEM of n observations, where n represents the number of animals or samples. For the kinetic studies one-way analysis of variance (ANOVA) and Bonferroni's test were used. For the study of the drug treatment effects on immunohistochemistry markers and lesion volume, Student's t-test was used. For the locomotor activity and body weight data, two-way (group and time post-injury) repeated-measures ANOVA was used, and intergroup differences were analyzed by ANOVA followed by Bonferroni's multiple comparison test at each corresponding time point. Statistical significance was set at p < 0.05 (Statview 5.0 software; Abacus Concept Inc., Berkeley, CA).
Results
Time course study of microglial activation and diffuse axonal injury after TBI
CD11b and ß-APP were used to evaluate the time course of post-TBI microglial activation and DAI, respectively.
CD11b-immunolabeled cells were not detected in naive mice (Fig. 1A), whereas they appeared in the contusional and peri-contusional areas of the right cerebral parenchyma after TBI. Their number was increased at 6 h post-TBI, and reached its maximum at 24 h, followed by a gradual decrease from 48 h until 7days post-TBI, when no more immunolabeled cells were detected (Fig. 1B).
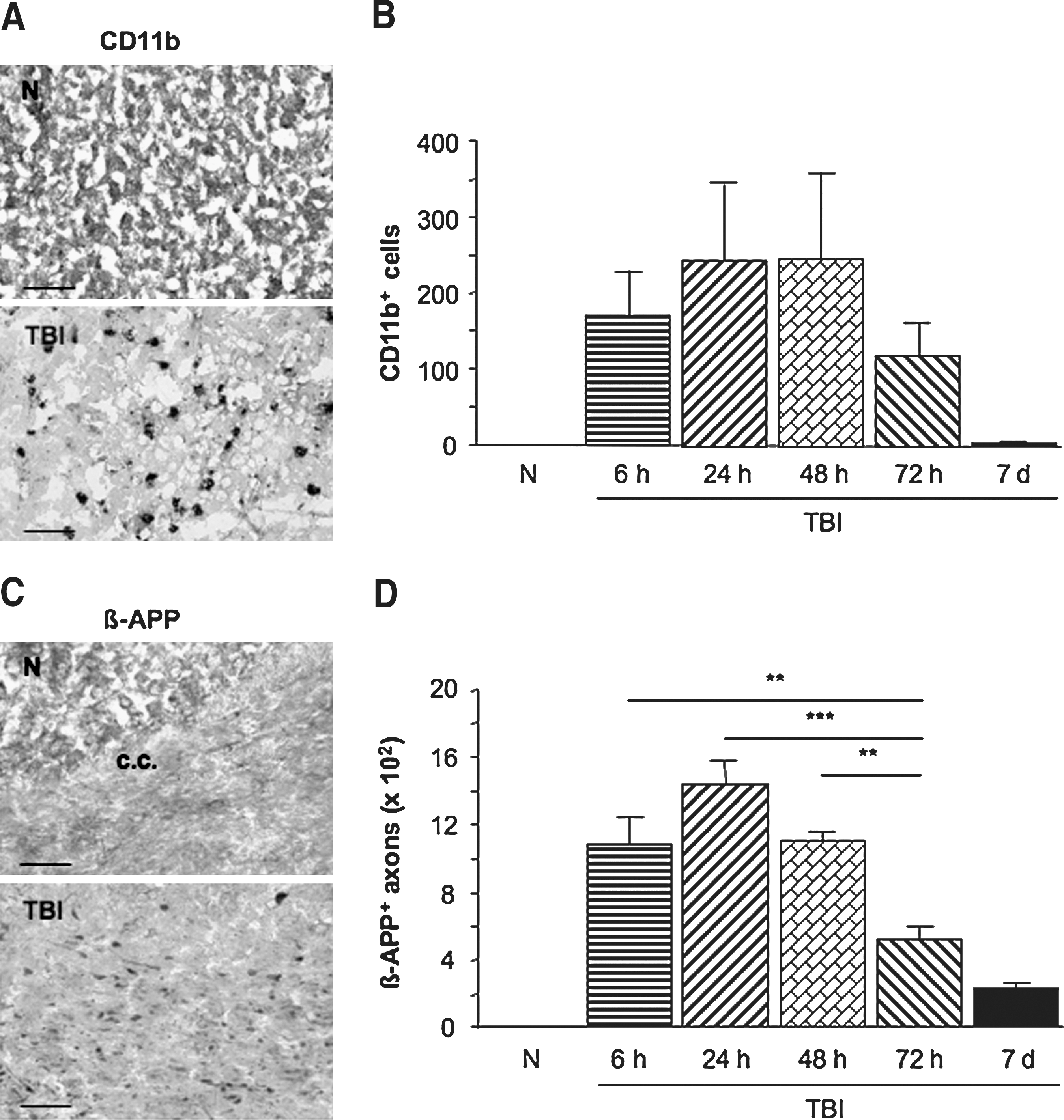
Evolution of cerebral CD11b-immunolabeled cells and axonal ß-amyloid precursor protein (ß-APP) accumulation as revealed by immunohistochemistry in naive (N) and in TBI mice 6, 24, 48, and 72 h, and 7 days post-TBI. (
ß-APP-immunolabeled axons were also not detected in naive mice, whereas they appeared as swollen axons and retraction bulbs in the corpus callosum after TBI (Fig. 1C and 1D). Their number increased at 6 h (p < 0.01) post-TBI, and continued to climb until 24 h (p < 0.001), followed by a gradual decrease from 48 h (p < 0.01) until 7days post-TBI (Fig. 1D).
Effects of minocycline treatment on the histological outcome after TBI
The effects of minocycline treatment have been tested on microglial activation, DAI, and the focal lesion at 24 h following TBI, when microglial activation and DAI reach their peak levels.
Numbers of CD11b-immunolabeled cells were markedly increased in the vehicle-treated TBI group (Fig. 2A). Minocycline treatment was able to reduce the post-TBI increase of CD11b immunolabeled cells by 59% (p < 0.05; Fig. 2A).
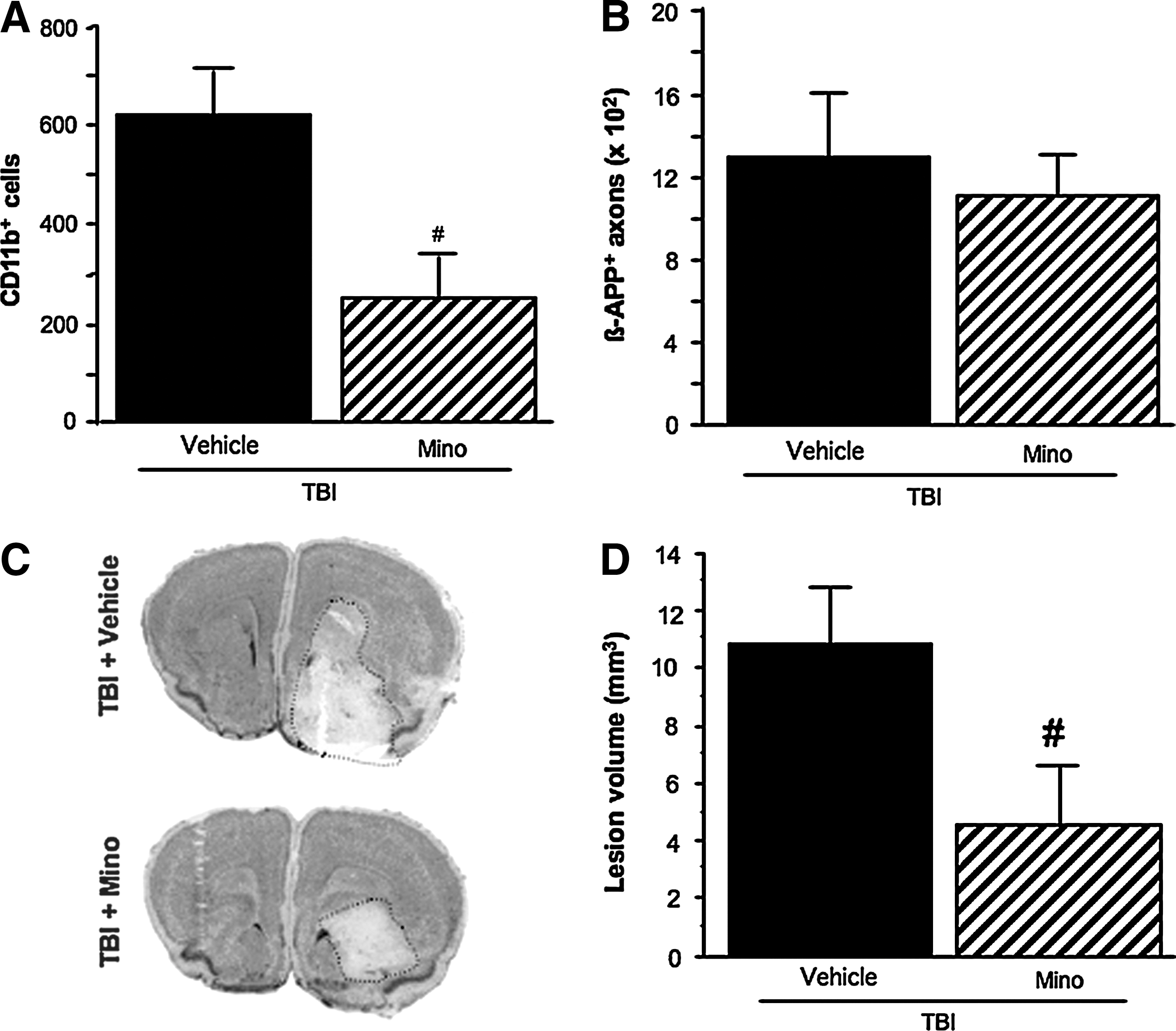
The effects of minocycline on cerebral CD11b-immunolabeled cells (
A marked increase in ß-APP-immunolabeled axons was observed in the vehicle-treated TBI group (Fig. 2B). Minocycline treatment did not alter the post-TBI increase in ß-APP immunolabeling (Fig. 2B).
A widespread lesion area was observed at the contrecoup-injured site of the brain (Fig. 2C), with a lesion volume of 10.8 ± 1.9 mm3 in the vehicle-treated TBI group (Fig. 2D). Minocycline markedly decreased the post-TBI lesion volume by 58% (p < 0.05; Fig. 2D).
Long-term effects of minocycline on TBI-induced locomotor hyperactivity
The effects of minocycline on TBI-induced changes in spontaneous horizontal (locomotion; Fig. 3A), and vertical (rearing; Fig. 3B) locomotor activity were followed for up to 12 weeks post-TBI. TBI induced persistent locomotor hyperactivity, with a significant increase in both horizontal and vertical activities from 2 days to 12 weeks following TBI. Although a slight decrease in hyperactivity was observed at 10 and 12 weeks, no significant changes were seen over time in the vehicle-treated TBI group. The treatment with minocycline led to a significant decrease in TBI-induced hyperactivity from 2 days until 8 weeks post-TBI (p < 0.05; Fig. 3).
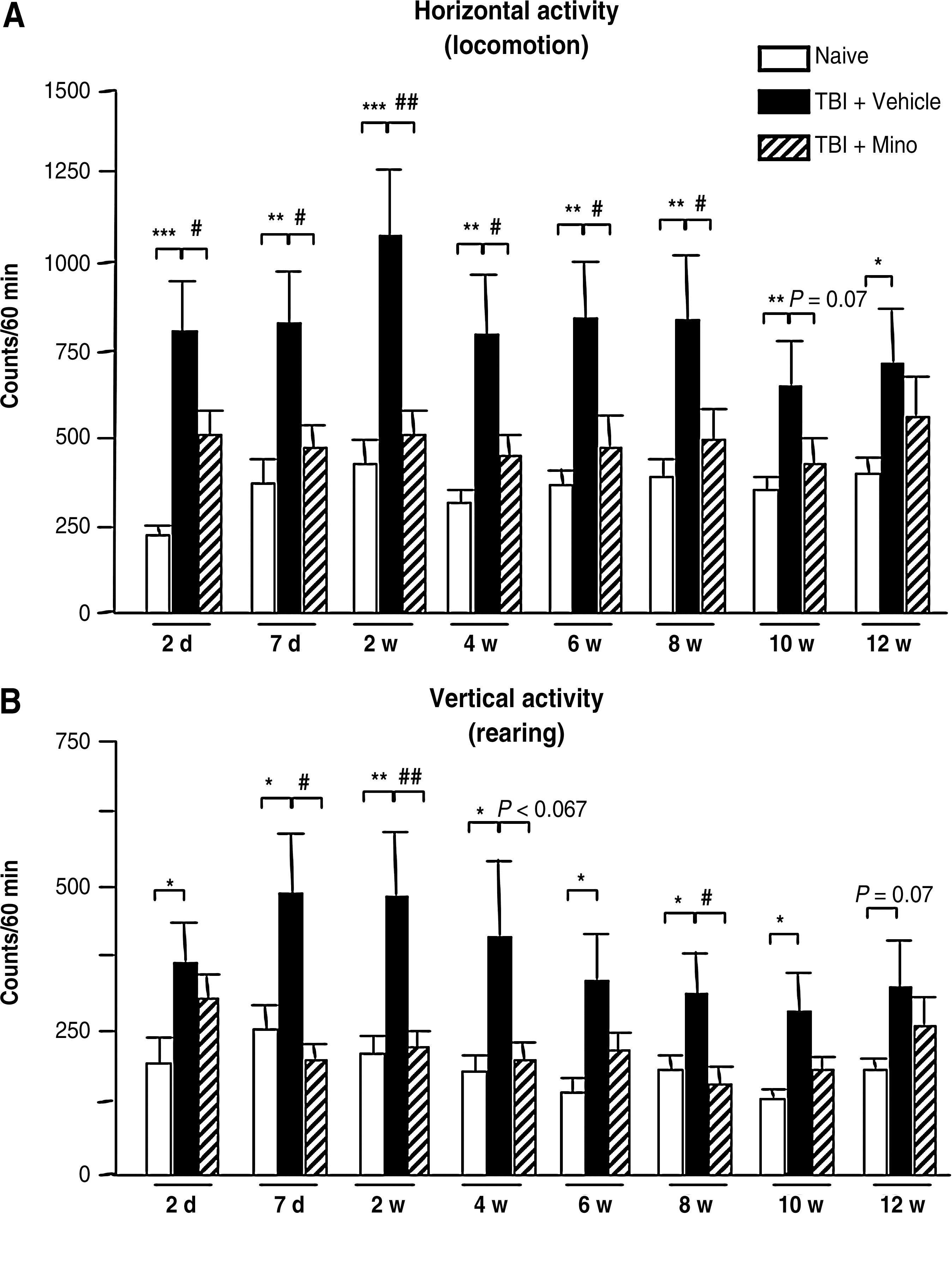
The long-term effects of minocycline on spontaneous horizontal (locomotion) (
Long-term effects of minocycline on TBI-induced body weight loss
A notable body weight loss was seen in both the vehicle-treated and minocycline-treated TBI groups at 2 days post-TBI compared with the naive group (p < 0.001; Fig. 4). Minocycline-treated TBI mice gained weight faster than vehicle-treated TBI mice from 7 days following TBI onward (p < 0.05), and reached a mean body weight close to that of naive mice at 4 weeks post-TBI. The difference in body weight between the vehicle-treated and minocycline-treated TBI groups increased gradually up to 12 weeks following TBI (p < 0.01; Fig. 4).

Graph showing the long-term effect of minocycline on body weight changes. The body weight was measured in naive mice, and in the two groups of TBI mice that received either the vehicle (Vehicle; phosphate-buffered saline 0.1 M IP, pH 7.4), or minocycline (Mino). Minocycline was administered at a dose of 90 mg/kg i.p. 5 min after TBI, followed by two other administrations (45 mg/kg i.p.) given at 3 and 9 h post-TBI. Measures were performed at different time points, including day zero (just before TBI), and 2 and 7 days and 2, 4, 6, 8, 10, and 12 weeks post-TBI. Data are expressed as the mean ± standard error of the mean (n = 10–12 per group), and were analyzed by two-way repeated-measures analysis of variance (ANOVA; for group p < 0.01; for time post-injury p < 0.001; for the interaction group x time post-injury p < 0.001). Intergroup differences were analyzed by ANOVA, followed by Bonferroni's multiple comparison test at each corresponding time point (**p < 0.01 and ***p < 0.001 versus naïve mice; # p < 0.05 and ## p < 0.01 versus vehicle-treated TBI mice; TBI traumatic brain injury).
Effects of minocycline on the spontaneous locomotor activity of naive mice
Treatment with minocycline did not significantly alter either the horizontal (p = 0.06) or the vertical (p = 0.13) spontaneous locomotor activity of naive mice compared to the vehicle-treated group (Fig. 5 A and B). Nevertheless, a trend towards an increase of horizontal locomotor activity was observed in minocycline-treated mice (P = 0.06).
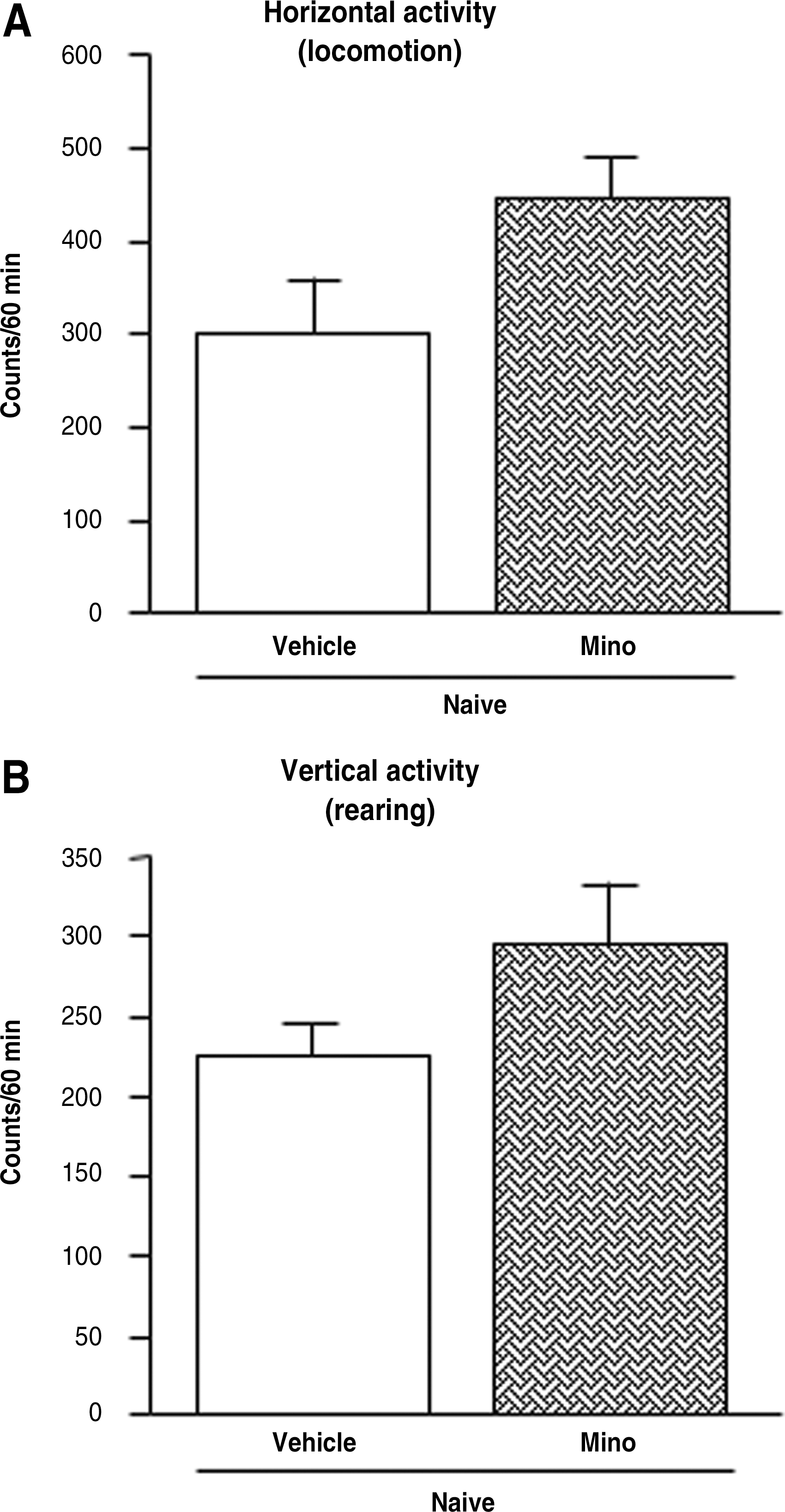
Minocycline's effects on the spontaneous locomotor activities of naive mice as evaluated by an automated actimeter. Minocycline's effects on total horizontal (locomotion) activity (
Discussion
Recently, we have shown the anti-edematous and anti-inflammatory effects of acute treatment with minocycline in a mouse model of closed head injury (Homsi et al., 2009), and the present study provides additional data on the beneficial effects of minocycline on histological outcome and locomotor hyperactivity following TBI. Minocycline is able to decrease the focal brain lesion and attenuate microglial activation after TBI. This neuroprotective effect of minocycline is not accompanied by a reduction in DAI. Additionally, this study provides new data regarding the long-term functional recovery occurring after minocycline administration.
First of all, kinetic studies have been undertaken to evaluate the microglial activation and DAI progression seen after TBI. An early increase in microglial/macrophage activation, as shown by an increase in CD11b immunolabeling, was seen in the contusional and peri-contusional areas from 6 h until 72 h post-TBI. A gradual decrease in CD11b immunolabeling was observed up to 7 days post-TBI, that may be due to a time-related attenuation of post-TBI microglial/macrophage activation, or to a switch of the expression of surface markers. In fact, microglia/macrophages express alternative surface markers over time (Guillemin and Brew, 2004; Kigerl et al., 2009), and therefore a reduction in one specific marker's expression does not necessary mean an attenuation of cell activation. Additionally, a post-TBI increase in microglial/macrophage activation has also been reported in humans and in different experimental models of TBI (Csuka et al., 2000; Kelley et al., 2007; Oehmichen et al., 1999). Furthermore, this TBI-induced microglial/macrophage activation was accompanied by a similar TBI-induced DAI profile (i.e., a rapid increase in axonal ß-APP immunolabeling in the corpus callosum from 6 h until 7 days post-TBI). These data provide evidence that our TBI model is suitable for the study of DAI following TBI, since axonal ß-APP accumulation is a hallmark of DAI (Gentleman et al., 1993; Sheriff et al., 1994; Stone et al., 2000). The time course of axonal ß-APP accumulation in our model is in accordance with that found in the fluid percussion TBI model in rats, which shows a significant increase in the accumulation of ß-APP in the corpus callosum from 1 day until 7 days post-TBI (Baker et al., 2002). Similarly, Ciallella and collaborators (2002), using the controlled cortical impact (CCI) TBI model in rats, found high ß-APP immunoreactivity levels at 1 and 3 days post-TBI in both the contused cortex and ipsilateral hippocampus, which declined at 14 days post-injury.
Results of the present study confirm the existence of a focal lesion after closed-head injury under our experimental conditions (Hellal et al., 2003). Treatment with minocycline induced a marked attenuation of the TBI-induced brain lesion, accompanied by a significant reduction in microglial activation. These effects are consistent with other findings seen in different models of focal TBI, and in a transient model of focal ischemia (Bye et al., 2007; Sanchez Mejia et al., 2001; Yrjanheikki et al., 1999), in which authors reported a diminution in both lesion volume and microglial activation in the peri-contusional area induced by minocycline. However, treatment with minocycline did not show any effect on the TBI-induced axonal accumulation of ß-APP seen in our study. This could be explained by the fact that TBI triggers a rapid and robust inflammatory response (i.e., a rapid release of TNF-α within 1 to 3 hours post-TBI from activated microglia), which may contribute to an early axonal accumulation of ß-APP (Kita et al., 2000). Thus, the attenuation of microglial activation by minocycline in our model either occurs too late to stop DAI progression, or it is insufficient to reduce the initiation of the inflammatory reaction and DAI progression. DAI may also occur independently of inflammation in this model of TBI. Taken together, the results of this study indicate that under our experimental conditions minocycline's capacity to reduce microglial activation may attenuate focal injury, but does not prevent DAI, at 24 h post-TBI.
It is worth noting that many neurological disorders and brain lesions, including TBI, can cause motor dysfunction such as locomotor hyperactivity (Li et al., 2006; Pullela et al., 2006; Viggiano, 2008). In this study, for the first time we used an automated actimeter to measure the spontaneous locomotor activity that occurs after TBI. We detected significant locomotor hyperactivity, from 2 days until 12 weeks post-TBI, which could be due to the severity of the injury and the site of the brain lesion, or may be caused by neurotransmitter changes that occur following insult (Fujinaka et al., 2003; Goldstein, 2006; Viggiano, 2008). Indeed, locomotor activity has been reported to increase due to lesions in the cerebral cortex, in the striatum, and in the olfactory bulbs (Viggiano, 2008), areas that are injured in our model. Furthermore, the increase in locomotor activity may also be due to the action of subcortical activating systems such as the dopaminergic system, as well as to the action of noradrenaline (Reid and Hamm, 2008; Viggiano, 2008). Another possible explanation for the hyperactive behavior is potential alterations in the two main neurotransmitter systems: the excitatory neurotransmitter glutamate, and the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Viggiano, 2008). The excitotoxic role of glutamate is well established following TBI. In fact, glutamate concentrations are elevated in both animals (Faden et al., 1989; Katayama et al., 1990) and humans (Baker et al., 1993; Bullock et al., 1998) following TBI. Other studies indicate that both ionotropic (NMDA) and metabotropic glutamate receptors are involved in the secondary processes occurring after TBI (Choi, 1988; Fei et al., 2007). Therefore, suppressing excessive post-TBI glutamatergic neurotransmissions or enhancing the inhibitory function of GABA neurotransmission could help establish a balance between these two systems and attenuate locomotor hyperactivity (Viggiano, 2008).
Furthermore, we endeavored to test the effect of minocycline on TBI-induced locomotor hyperactivity. For the first time we found that minocycline significantly reduced both horizontal and vertical activities for up to 8 weeks in our TBI model. This may be explained by minocycline's beneficial effect on focal lesion volume, or may be due to the ability of minocycline to mitigate excessive glutamate release and to reduce glutamatergic system activation both in vitro (González et al., 2007; Tikka and Koistinaho, 2001) and in vivo (González et al., 2007; Manev and Manev, 2009; Zhang et al., 2007). It is worth noting that minocycline's attenuating effect on locomotor hyperactivity was not due to its ability to reduce baseline locomotor activity, since no attenuation was observed in naive mice in the present study. We even observed a trend toward an increase in basal locomotor activity that may be caused by minocycline, which could be due to its irritant properties (Fagan et al., 2004). Additionally, the beneficial effect of minocycline on locomotor function following TBI is accompanied by a gradual recovery from post-TBI body-weight loss. In fact, a significant body-weight loss was seen after TBI at 2 days post-insult, and minocycline was able to reduce it significantly, that nearly allowed the animals to attain the same body-weight as naive mice at 4 weeks post-TBI. Thus minocycline is able to reduce TBI-induced body-weight loss and locomotor hyperactivity over the long term.
In conclusion, results of the present study confirm early microglial activation following TBI, and provide evidence for the existence of both diffuse and focal injuries in our TBI model. For the first time, this study provides evidence of long-lasting locomotor hyperactivity after TBI. Additionally, acute treatment with minocycline was able not only to reduce microglial activation and focal injury, but it also had a long-lasting attenuating effect on locomotor hyperactivity after TBI. Taken together, under these experimental conditions, microglial activation seems to play an important role in the development of TBI-induced focal injury and the subsequent locomotor hyperactivity, and its acute inhibition with minocycline provides long-lasting functional recovery. The findings detailed here indicate that acute treatment with minocycline may represent a promising new therapeutic strategy for head-injured patients.
Footnotes
Acknowledgments
The authors would like to thank Dr. B. Saubaméa and V. Mignon (Cellular and Molecular Imaging Platform, Paris Descartes University, UFR Pharmacy) for their assistance in the imaging facility.
Author Disclosure Statement
No competing financial interests exist.