
Abstract
Gap junctions are conductive channels formed by membrane proteins termed connexins (Cx), which permit the intercellular exchange of metabolites, ions, and small molecules. Junctional permeability is regulated by pH, membrane potential, and intracellular secondary messengers. The purpose of this study was to elucidate the expression and distribution of astrocytic gap junctions in the hippocampus and the cortex after traumatic brain injury (TBI) in vivo. Adult male Sprague-Dawley rats (300–400 g) were subjected to lateral fluid percussion injury (FPI) at moderate severity (2.6–2.8 atm, 12 msec) using a Dragonfly device model. Phosphorylated gap junction protein levels were quantified using Western blot analysis. Spatial distribution of immunoreactivity for phosphorylated Cx43 (p-Cx43) was analyzed by immunohistochemistry. Our findings showed that p-Cx43 expression in the ipsilateral hippocampus was significantly induced at 1 h after TBI, and remained at a high level until 24 h after injury. The p-Cx43 protein content reached a maximum level at 6 h after injury. In addition, the immunoreactivity for p-Cx43 was localized in the astrocytes surrounding ipsilateral CA3 pyramidal neurons. On the other hand, the protein level in the ipsilateral cortex was not significantly different at any time point after TBI. Double immunostaining using phosphorylated ERK (p-ERK) showed that p-Cx43 and p-ERK immunoreactivities were enhanced in the same astrocytes at 6 h after injury. These findings suggest that astrocytic gap junctions participate in pathophysiological processes in the hippocampus after TBI.
Introduction
Gap junctions are conductive channels that mediate the direct cytoplasmic exchange of small hydrophilic molecules and ions, such as Ca2+, ATP, and inositol triphosphate, between adjacent cells (Bennett et al., 1991; Giaume and McCarthy, 1996), and they are considered to be critical for tissue homeostasis (Dermietzel, 1998). Recent studies have indicated that several connexins (Cxs), namely gap junction proteins, are expressed by various cells in the central nervous system (CNS). In particular, Cx43 and Cx30 are expressed predominantly in astrocytes; Cx26, Cx32, and Cx36 are synthesized in neurons; and Cx29, Cx32, and Cx47 are localized in oligodendrocytes (Bruzzone and Ressot, 1997; Dermietzel, 1998; Teubner et al., 2001). In addition, the intracellular condition and phosphorylation state of Cx affects intercellular permeability via gap junctions. Phosphorylation of Cx43 by the ERK signaling pathway induces the reduction of intercellular permeability via gap junctions (Brandes et al., 2002; Cottrell et al., 2003).
Recent studies have demonstrated that Cx43 is greatly enhanced in astrocytes in the selectively vulnerable CA1–CA2 region, which correlates with excessive selective vulnerability to cerebral ischemia (Rami et al., 2001), and global ischemia induces selective upregulation of Cx32 and Cx36 in vulnerable CA1 neurons before the onset of neuronal death (Oguro et al., 2001). However, Cx expression and distribution following TBI in vivo have not been determined. In the present study, we investigated the expression of phosphorylated Cx43 (p-Cx43) immunoreactivity in the rat hippocampus and cortex after experimental TBI. As a result, temporal and spatial expression of p-Cx43 immunoreactivity was observed in the hippocampus but not in the cortex after TBI.
Methods
Surgical procedures
The lateral FPI model has been demonstrated previously (McIntosh et al., 1989). A total of 88 adult male Sprague-Dawley rats (10–11 weeks old; weight 350–400 g) were housed in individual cages under controlled environmental conditions (12/12-h light-dark cycle at room temperature), with food and water available, for at least 1 week before surgery. On day 1, the rats were anesthetized with sodium pentobarbital (50 mg/kg IP) and mounted in a stereotaxic frame. A 4.8-mm craniectomy was performed over the right parietal bone (centered at 4.0 mm posterior and 3.0 mm lateral to the bregma). The dura was left intact during the procedure. A plastic Luer-Lok fitting was cemented over the craniectomy site with dental cement.
On the following day, the rats were anesthetized with isoflurane in a 2:1 mixture of nitrous oxide and oxygen, a polyethylene catheter was inserted into the femoral artery, intubation was performed using a 14-gauge angiocatheter, and then they were mechanically ventilated with 2–2.2% isoflurane. The rectal temperature (37.0–37.5°C) was assessed with a rectal probe and maintained within a normal level using a heating pad. Arterial blood samples were analyzed intermittently (pre-injury and at 5 and 15 min after injury), and blood pressure was measured continuously. The rats were subjected to FPI at a moderate severity (2.6–2.8 atm, 12 msec) using a Dragonfly fluid percussion device (HPD-1700; Dragonfly R&D, Silver Spring, MD). The pulse pressure was measured extracranially with a pressure transducer (211 B4; Kistler Instrument Corp., Amherst, NY), and then recorded using a digital storage computer (MacLab; AD Instruments, Bella Vista, N.S.W., Australia). Sham control animals were subjected to the same procedures except for the actual insult.
All procedures were approved by the Animal Care and Use Committee of National Defense Medical College.
Tissue preparation
For Western blot analysis, the rats were euthanized by decapitation under IP anesthesia with pentobarbital sodium at 5 min, 30 min, 60 min, 6 h, 24 h, and 72 h after injury (n = 4 per each time point). Both cortical and hippocampal tissue specimens were immediately excised and submerged in ice-cold artificial cerebrospinal fluid (containing 0.6 mmol/L NaH2PO4/2H2O, 3.35 mmol/L KCl, 138.6 mmol/L NaCl, 9.9 mmol/L dextrose, 21 mmol/L NaHCO3, 2.5 mmol/L CaCl2, and 1 mmol/L MgCl2). The tissue specimens were aliquoted, frozen, and kept at −80°C until analysis. Sham control rats were sacrificed at 6 h after surgery (n = 4). For immunohistochemical analysis, the rats were perfused transcardially with normal saline followed by 4% buffered paraformaldehyde under IP anesthesia with pentobarbital sodium at the same time points after injury (n = 4–5 per each time point). The brains were removed and embedded in paraffin after fixation in 4% buffered paraformaldehyde, followed by 0.1 mmol/L phosphate-buffered saline (PBS; pH 7.4) for 24 h at 4°C, and then serial coronal sections (5 μm) were prepared. The existence of morphological changes, such as gliding contusional hemorrhage, was confirmed in all FPI animal sections.
Western blot analysis
Cortical and hippocampal tissues were used for Western blot analysis. The frozen tissue specimens were homogenized with ice-cold histolysis buffer [50 mmol/L HEPES-KOH (pH 7.5), 250 mmol/L NaCl, 1 mmol/L EDTA, 1% Naridet P-40, 2 mmol/L Na3Vo4, and protease inhibitor cocktail set III (no. 539134; Calbiochem, Darmstadt, Germany)]. The protein content was determined with a detergent-compatible protein assay reagent kit (Bio-Rad, Hercules, CA). Equal amounts of protein (200 μg) were loaded in each lane on a 12% Tris polyacrylamide gel for sodium dodecyl sulfate-polyacrylamide gel electrophoresis with loading buffer (100 mmol/L Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate, 20% glycerol, 0.2% bromophenol blue, and 200 mmol/L dithiothreitol). Protein was loaded without dithiothreitol when electrophoresis was performed under non-reductive conditions. The protein was transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Buckinghamshire, U.K.) at 250 mA for 2 h. The membranes were washed with Tris-buffered saline containing 0.05% Tween-20 (TTBS; pH 7.4), blocked with a 4% Block Ace (DS Pharma Biomedical Co., Osaka, Japan), and then incubated overnight at 4°C with either monoclonal total Cx43 antibody (1:8000, #C8093; Sigma-Aldrich, St. Louis, MO), polyclonal p-Cx43 antibody (1:100, #SC-101660; Santa Cruz Biotechnology, Santa Cruz, CA), or β-tubulin antibody (1:200, #SC-5274; Santa Cruz Biotechnology). The membranes were then incubated for 1 h at room temperature with horseradish peroxidase-linked anti-rabbit IgG secondary antibody (1:5000; Santa Cruz Biotechnology). Protein bands were detected by electrochemiluminescent (ECL) Western blotting detection reagent (GE Healthcare).
Immunohistochemistry
Immunohistochemistry was performed by the Universal Immuno-peroxidase Polymer (UIP) method. N-Histofine Simple Stain Rat MAX-PO (MULTI; Nichirei Co., Tokyo, Japan) was used for immunostaining by monoclonal anti-total Cx43 antibody and polyclonal anti-p-Cx43 antibody. After deparaffinization and hydration, sections in 10 mmol/L sodium citrate (pH 6.0) were boiled for 40 min at 98°C. Endogenous peroxidase was inactivated and blocked with 3% hydrogen peroxide for 10 min at room temperature and rinsed in PBS. The sections were incubated overnight at 4°C with anti-p-Cx43 antibody (1:50), then rinsed with 10 mmol/L PBS with Tween-20 (PBST). The sections were incubated with N-Histofine Simple Stain Rat MAX-PO (MULTI) at room temperature for 45 min. The sections were then rinsed with 10 mmol/L PBST twice and then PBS. Peroxidase activity was measured with diaminobenzidine (DAB). For immunohistochemistry of the total Cx43, the sections were incubated in 0.1% trypsin for 7 min at 37°C. After endogenous peroxidase was blocked, the sections were incubated for 1 h at 37°C with primary antibody against the total Cx43 (1:500). Thereafter, the sections were incubated with N-Histofine Simple Stain Rat MAX-PO (MULTI) at room temperature for 30 min. All other procedures were performed as described above. To evaluate morphological changes, adjacent sections were counterstained with hematoxylin.
Double immunostaining
The paraffin-embedded serial sections were double immunostained with p-ERK and p-Cx43, with a Dako AP universal kit (Dako, Kyoto, Japan) for p-Cx43, and the N-Histofine Simple Stain Rat MAX-PO (MULTI) for p-ERK. The sections were boiled at 95°C in citrate buffer (pH 6.0) for 10 min, and then incubated with polyclonal antibody against p-ERK (#9101; Cell Signaling, Danvers, MA) overnight at 4°C, followed by N-Histofine Simple Stain Rat MAX-PO (MULTI) at room temperature for 30 min. After p-ERK immunostaining was reacted with DAB, p-Cx43 immunostaining was reacted with a fuchsin substrate chromogen system (Dako). Co-localization of p-ERK (brown) and p-Cx43 (red) was identified by a pinkish-brown mixed color. Furthermore, we performed double immunostaining for GFAP and p-Cx43. After p-Cx43 immunostaining reacted with DAB, the sections were boiled at 95–100°C in citrate buffer (pH 6.0) for 10 min, and then incubated with pre-diluted rabbit anti-GFAP antibody (Dako) at room temperature for 1 h. Staining color was developed with a fuchsin substrate chromogen system (Dako).
Statistical analysis
All data are expressed as mean ± SD. The physiological parameters were analyzed by one-way repeated-measures analysis of variance (ANOVA). The densities of the immunoblot bands were quantified using Image J software (version 1.33u; National Institutes of Health, Bethesda, MD). Significant differences in Western blots were analyzed using the Kruskal-Wallis H test, followed by a post-hoc Mann-Whitney U test with Bonferroni correction. The level p < 0.05 was considered to be statistically significant.
Results
Physiological parameters were all within normal ranges during the surgical procedures, and no significant differences were found between the TBI and sham control groups after injury (Table 1).
The values are means ± standard deviation. The physiological parameters were all within normal ranges, and no significant differences were found between TBI and sham control rats after injury.
MABP, mean arterial blood pressure; Temp, rectal temperature; TBI, traumatic brain injury; P
The results for p-Cx43 and the total amount of Cx43 protein content are shown in Figure 1. A few differences in immunoreactivity for p-Cx43 were observed between the hippocampus and cortex in response to TBI. The immunoreactivity for p-Cx43 in the ipsilateral hippocampus was significantly induced at 1 h after injury (230.5 ± 38.2%; p < 0.05), persisted at a high level until 24 h after injury (216.5 ± 39.7%; p < 0.05), and thereafter gradually decreased (Fig. 1C). In addition, the p-Cx43 protein content reached a maximum level (283.5 ± 84.6%; p < 0.001) at 6 h after injury (Fig. 1C). In contrast, no significant change in the total amount of Cx43 in the hippocampus was seen after injury (Fig. 1B). These findings indicate that the induction of p-Cx43 after TBI might not be attributable to changes in the total amount of Cx43 protein. In the ipsilateral cortex site of injury, there were no significant differences in both the total amount of Cx43 and p-Cx43 at any time point post-TBI (Fig. 1E and F).

Time course of p-Cx43 and the total amount of Cx43 expression in the ipsilateral hippocampus (
The immunohistochemical results were consistent with those of Western blot analysis (Figs. 2 and 3). A prominent change in p-Cx43 immunoreactivity was observed in the ipsilateral hippocampus (Fig. 2). The immunoreactivity for p-Cx43 was greatly enhanced in the hippocampal CA3 region at 6 h after injury (Fig. 2C and D), and thereafter gradually reduced. Furthermore, the induction of p-Cx43 expression was localized in astrocytes surrounding ipsilateral CA3 pyramidal neurons (Fig. 2H). In contrast, immunoreactivity for p-Cx43 in the contralateral region was similar to that seen in the sham-injured rats at all time points post-injury (data not shown). A few immunopositive-stained cells for total Cx43 were detected in the sham-injured rats (Fig. 3A, E, and F). The expression of total Cx43 immunoreactivity was recognized not only in the vicinity of hippocampal pyramidal neurons, but also in distant regions, until 72 h after the procedure (Fig. 3D). In the ipsilateral cortex site of injury, the induction of total Cx43 expression was enhanced surrounding the gliding contusional region at 72 h after injury compared to sham control rats (Fig. 3H).
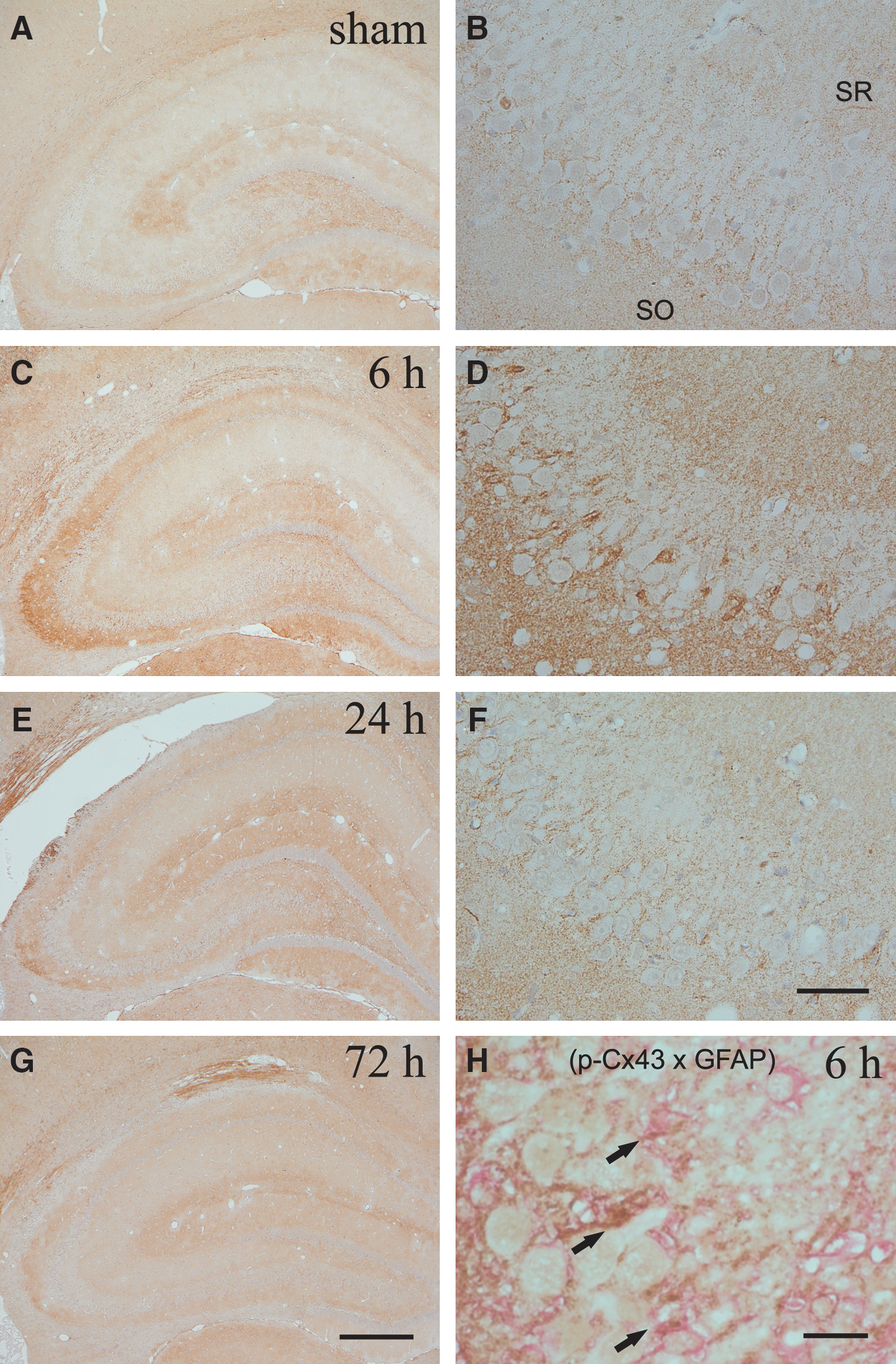
Photomicrographs show the spatial distribution of immunoreactivity for p-Cx43 in the ipsilateral hippocampus following TBI. Sections were prepared from the rats at 6 h (
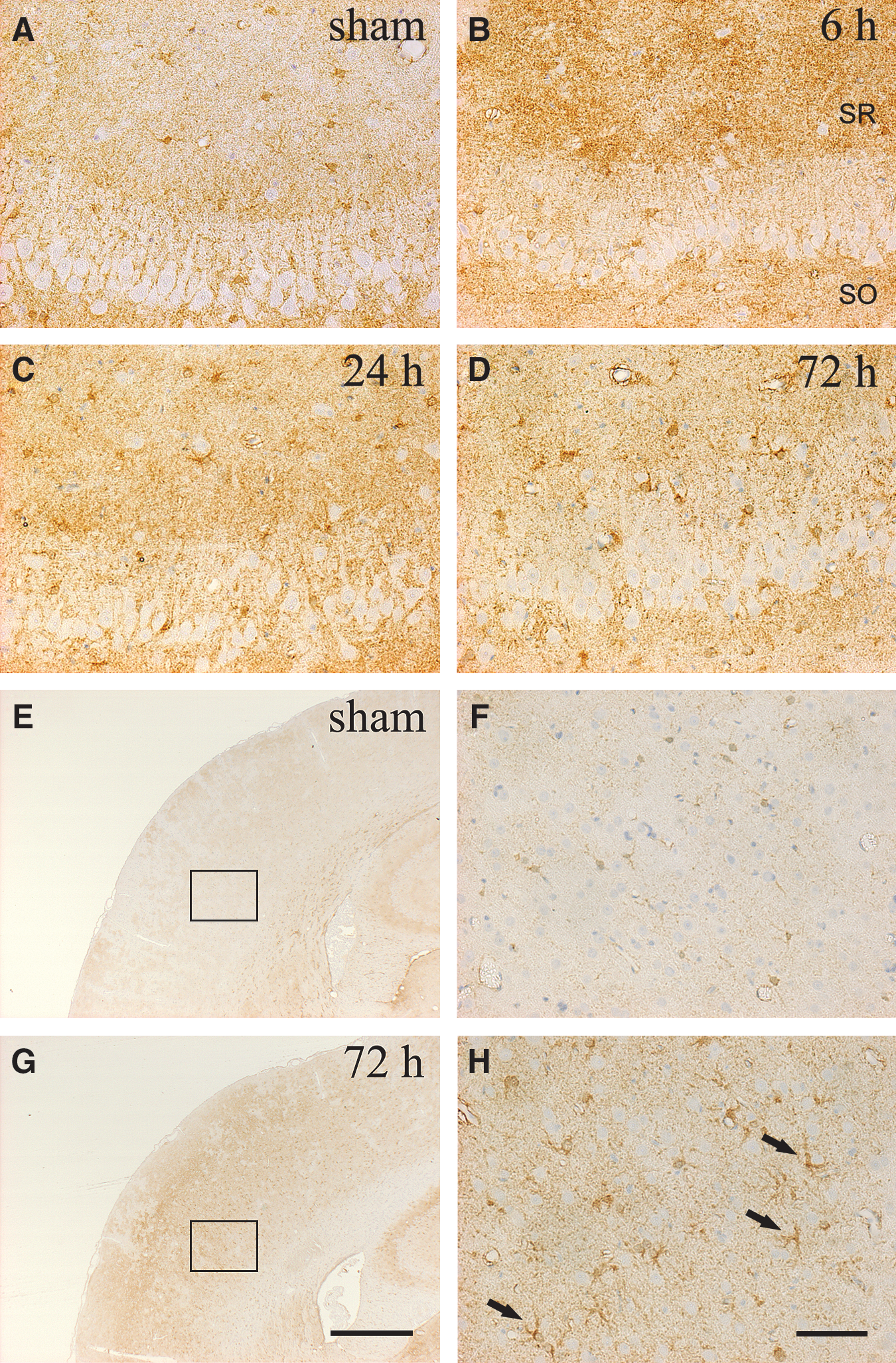
The photomicrographs show the distribution of immunoreactivity for total Cx43 in the ipsilateral hippocampus (
Double immunoreactivity for p-Cx43 and p-ERK clarified the alterations seen in these two proteins for astrocytes in the ipsilateral CA3 region after TBI (Fig. 4). Neither p-Cx43 nor p-ERK immunoreactivity was observed in sham control rats (Fig. 4A). In contrast, double immunoreactivity for p-Cx43 and p-ERK was enhanced in the same astrocytes surrounding the ipsilateral CA3 pyramidal neurons at 6 h after injury (Fig. 4C and D).
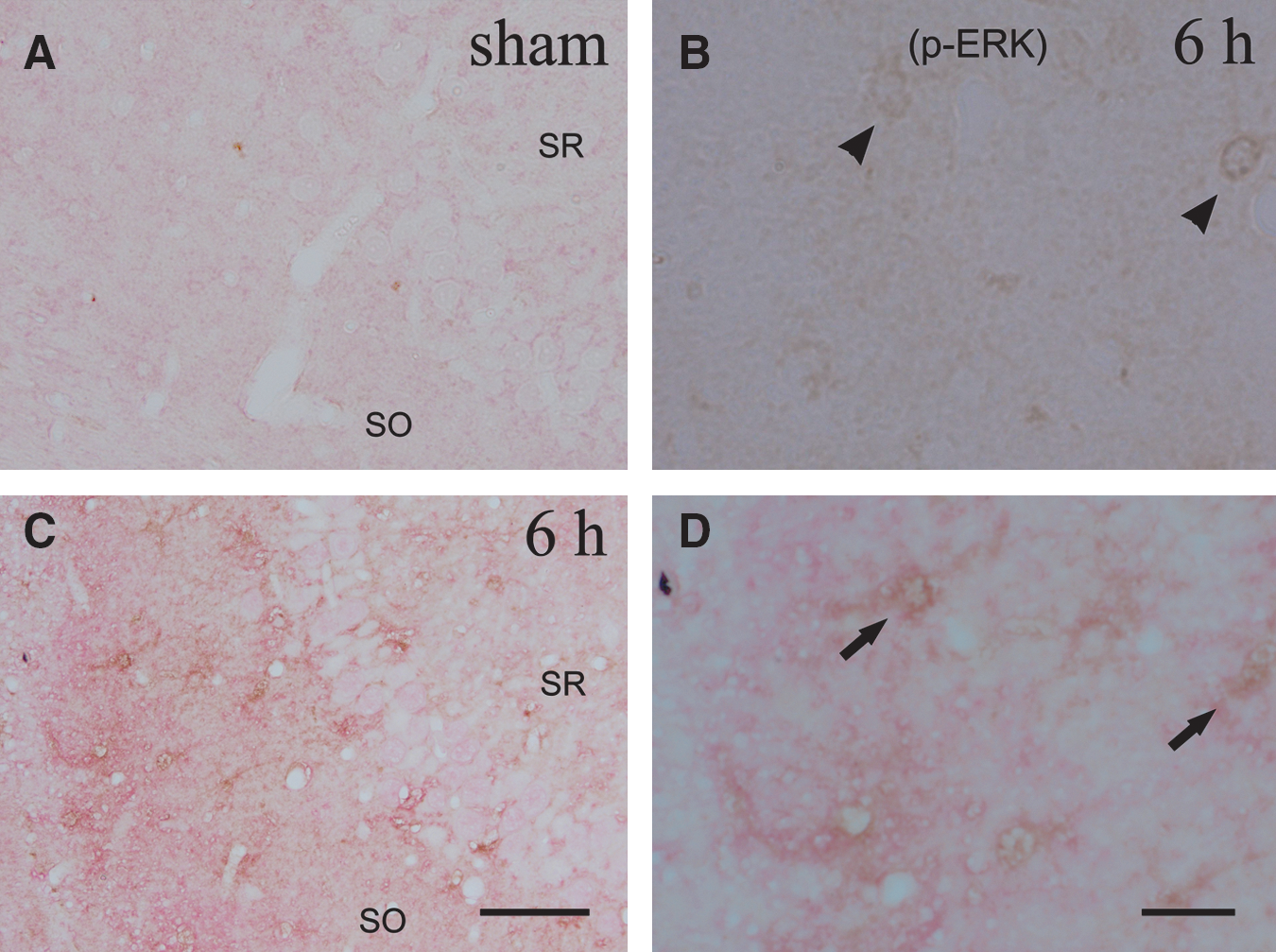
Photomicrographs show the cellular localization and time course of immunoreactivity for p-ERK (brown) and p-Cx43 (red) in the ipsilateral CA3 region. The sections were prepared from the rats at 6 h (
Discussion
Cx regulation after brain injury has been studied in various pathophysiological situations. Intercellular signal transduction through gap junctions contributes to secondary brain damage upon ischemic injury (Frantseva et al., 2002a; Hossmann, 1994; Lin et al., 1998). In addition, stress factors that pass through gap junctions may include apoptotic factors. Frantseva and associates (2002a) found that cell death is reduced in sections that were treated with antisense oligonucleotides for Cx43 after hypoxic-hypoglycemic insult. As such, a partial decrease in neuronal or glial gap junction communication is sufficient to reduce secondary damage. Lin and colleagues (1998) demonstrated that astrocytic gap junctions propagate transcellular signals, which exacerbate cell injury induced by calcium overload, oxidative stress, and metabolic inhibition in cerebral ischemia. Therefore, the authors suggested that dying glial cells killed neighboring cells that would otherwise have escaped injury; this process of glial “fratricide” may provide a basis for the propagation of secondary brain damage. Furthermore, Rami and co-workers (2001) have shown that inhibition of gap junction permeability effectively decreases neuronal death. On the other hand, heterozygotic Cx43 null (Cx43+/−) mice, which have significantly reduced Cx43 expression, show larger infarct lesions in comparison to wild-type (Cx43+/+) mice (Siushansian et al., 2001). Likewise, astrogliosis in the cortical ischemic lesion decreases significantly more in Cx43+/− mice than in Cx43+/+ mice, and caspase-3 staining in the penumbral lesion is stronger in Cx43+/− mice (Nakase et al., 2003). Therefore, the authors also suggested that astrocytic gap junctions contribute to the development of a reactive astrocytic network and the removal of cytotoxic factors.
Previous studies have indicated that spreading depression occurs after TBI or ischemic insult that propagates both neuronal and glial depolarization, and thereafter tends to induce neuronal damage (Katayama et al., 1990; Nedergaard and Hansen, 1993). Frantseva and associates (2002b) demonstrated that CNS injury decreases significantly in sections from Cx43 knockout mice compared to wild-type mice, using in vitro trauma models. The authors also suggested that gap junctions cause cellular vulnerability to injury while also amplifying excitotoxic components that represent an important contribution to the damage. However, the mechanisms of gap junction communication and selective vulnerability in the hippocampus following TBI have not been amply elucidated, and no reports have focused on astrocytic Cx expression after TBI in vivo. In the present study, astrocytic gap junction proteins were phosphorylated in the ipsilateral hippocampus after FPI. Immunoreactivity for p-Cx43 increased transiently in astrocytes of the selectively vulnerable CA3 region following TBI, whereas any obvious alteration of p-Cx43 expression was not detected in the CA1 subfield and contralateral hippocampus. These findings suggested that phosphorylation of Cx43 induces cell injury in the post-traumatic vulnerable region. Therefore, we speculated that phosphorylation of Cx43 may contribute to hippocampal dysfunction through astrocytic gap junction communication in the early phase after closed brain injury. On the other hand, immunoreactivity of the total amount of Cx43 was unchanged during the time course after TBI. These findings suggested that TBI induces transient phosphorylation of Cx43 in the hippocampus; however, this phenomenon may not be related to the alteration of the total amount of Cx43.
Reactive astrocytes are known to be the most prominent response to TBI. Dietrich and colleagues (1999) discovered that TBI induces GFAP gene expression that might be a sensitive molecular marker for evaluating the global response in progressive glial scarring in the rat brain. Reactive astrocytes have a beneficial effect on neuronal survival and on repair of the damaged blood–brain barrier (Bush et al., 1999). On the other hand, Mandell and co-workers (2001) suggested that astroglial reactions, which lead to reactive astrogliosis, have both beneficial and detrimental consequences for functional recovery of neurons. Glial scars may impede any neuronal repair and axonal regeneration after an injury to CNS; however, reactive astrocytes might support axonal regeneration (Ridet et al., 1997). The present results show that total Cx43 immunoreactivity was localized in astrocytes in the vicinity of the gliding contusion in the ipsilateral cortex at 72 h after TBI. These findings suggested that the induction of Cx43 expression is a substantial factor in the astroglial reaction, and potentiates the intercellular signal transduction via gap junction after injury. Therefore, we speculated that astrocytic gap junctions might play an important role, such as having a beneficial effect in restoring neuronal damage and maintaining the CNS through their gap junctions during the late phase after injury.
In addition, a number of endogenous compounds have been shown to regulate gap junction communication; therefore, neurotransmitter and intercellular signals can control junctional permeability (Giaume and McCarthy, 1996; Perez-Velazquez et al., 1994; Rouach et al., 2000). The activation of glutamate receptors has the opposite effect on gap junctions by inducing intercellular Ca2+ signaling (Enkvist and McCarthy, 1994; Giaume and Venance, 1998; Muller and Kettenmann, 1995). On the other hand, the induction of Cx43 phosphorylation by MAPK decreases intercellular permeability via gap junctions. Zvalova and associates (2004) suggested that p38/stress-activated protein kinase-2 is a central mediator of IL-1β-induced rapid gap junction closure in cultured mouse astrocytes. In addition, Cx43 phosphorylation at tyrosine residues may regulate gap junctional coupling, as the phenomenon could contribute to the downregulation of gap junction coupling in astrocytes during ischemia (Li et al., 2005). A previous study has shown that the induction of p-ERK expression is observed in astrocytes surrounding CA3 neurons at 6 h after TBI (Otani et al., 2002). Then the activation of ERK 1/2 induces phosphorylation of Cx43 and inhibition of gap junction communication in endothelial cells (Brandes et al., 2002). On the other hand, double immunostaining results in the present experiments showed that p-ERK immunoreactivity is expressed in astrocytes surrounding the pyramidal CA3 region in which p-Cx43 immunoreactivity is also observed. These findings suggest a complex relationship between Cx43 and ERK1/2 phosphorylation. It is well known that post-traumatic amnesia is a common feature during the acute phase after TBI, and it seems to be related to hippocampal dysfunction (Scheff et al., 1997). We speculated that astrocytic gap junction communication may be suppressed transiently by ERK phosphorylation in the hippocampus after injury, and that partial pathological hippocampal dysfunction, such as post-traumatic amnesia and post-traumatic stress disorder, occur by reduction of cell-cell permeability through gap junctions following TBI.
We demonstrated for the first time an alteration of p-Cx43 immunoreactivity following lateral FPI in the rat brain. Our data suggest that phosphorylation of Cx43 in the hippocampus, but not in the cortex, is associated with molecular sequelae of TBI, and that differential Cx expression reflects the roles of astrocytic gap junctions in the selectively vulnerable CA3 region. Alternatively, astrocytic gap junctions might regulate the recovery process from contusional brain injury in the cortex. Thus astrocytic gap junctions may play a critical role in the regulation of tissue homeostasis, and may be a crucial configuration factor of the cell membrane to rectify hippocampal function following TBI. Astrocytic gap junctions may be an important molecular target to elucidate the mechanism of neuronal and astroglial damage after TBI. However, the mechanism by which gap junctions function after TBI is still not yet fully understood. Further investigation, such as using specific inhibitors or a fluorescent dye diffusion technique, is necessary to clarify the relationship between gap junctions and intercellular signaling following TBI.
Footnotes
Acknowledgments
We thank Akiko Yano, Namiko Nomura, and Dr. Takamoto Suzuki for their excellent technical assistance, and Drs. Hiroshi Kato, Shinichiro Ishihara, Nobusuke Tsuzuki, Akira Ohnuki, and Takahito Miyazawa for their valuable help in preparing this manuscript.
Author Disclosure Statement
No competing financial interests exist.