
Abstract
A hallmark of severe traumatic brain injury (TBI) is the development of post-traumatic epilepsy (PTE). However, the mechanisms underlying PTE remain poorly understood. In this study, we used a controlled cortical impact (CCI) model in rats to examine post-traumatic changes in neocortical excitability. Neocortical slices were prepared from rats at 7–9 days (week 1) and 14–16 days (week 2) after CCI injury. By week 2, we observed a substantial gray matter lesion with a cavity that extended to the hippocampal structure. Fluoro-Jade B staining of slices revealed active neuronal degeneration during weeks 1 and 2. Intracellular and extracellular recordings obtained from layer V revealed evoked and spontaneous epileptiform discharges in neocortices of CCI-injured rats. At week 1, intracellular recordings from pyramidal cells revealed evoked epileptiform firing that was synchronized with population events recorded extracellularly, suggestive of increased excitability. This activity was characterized by bursts of action potentials that were followed by recurrent, repetitive after-discharges. At week 2, both spontaneous and evoked epileptiform firing were recorded in slices from injured rats. The evoked discharges resembled those observed at week 1, but with longer burst durations. Spontaneous activity included prolonged, ictal-like discharges lasting up to 8–10 sec, and briefer interictal-like burst events (<1 sec). These results indicate that during the first 2 weeks following severe CCI injury, there is a progressive development of neocortical hyperexcitability that ultimately leads to spontaneous epileptiform firing, suggesting a rapid epileptogenic process.
Introduction
T
One approach that has been used extensively to study post-traumatic epileptogenesis in the neocortex involves partial isolation of neocortical circuits by surgical undercut to simulate deafferentation and white matter (axonal) damage caused by TBI (Hoffman et al., 1994; Prince and Tseng, 1993). In this model, evoked and spontaneous epileptiform events, particularly those resembling interictal-like discharges, may develop within 2–3 weeks after the injury. A second approach involves neocortical implantation of blood or blood products, such as ferrous chloride, to replicate some of the pathologies of TBI, such as intraparenchymal hemorrhage (Hammond et al., 1980). Previous investigations conducted by our laboratory have utilized an in vitro model of neocortical trauma, in which acute rat brain slices are mechanically injured via surgical removal of the superficial cortical laminae, which leads to persistent epileptiform activity 1–2 h after trauma (Yang and Benardo, 1997; Yang et al., 2007). However, while useful, these animal models all lack one or more features of TBI in humans, such as diffuse grey matter lesions, a unique focus, hemorrhagic components (Yablon and Dostrow, 2007), and notably, the generation of spontaneous seizure-like epileptiform discharges in cortical circuits.
CCI injury is a widely used model of TBI that has been applied to studies of trauma-induced cellular and molecular pathologies (Lighthall, 1988), cognitive disruptions (Saatman et al., 2006), motor dysfunction (Cernak, 2005), and TLE (Hunt et al., 2009). However, CCI has not yet been studied as a model for post-traumatic neocortical epilepsy. Although acute post-traumatic seizures have been reported with CCI in the immediate post-injury phase (Nilsson et al., 1994), no studies have yet examined persistent, chronic changes in neocortical excitability following CCI injury. In this study, we investigated whether CCI leads to neocortical epileptogenesis in rats. Animals were subjected to severe CCI neurotrauma, and then assessed for changes in cortical circuit excitability at 1 and 2 weeks following injury.
Methods
CCI injury
Under a protocol approved by the Institutional Animal Care and Use Committee of SUNY Downstate Medical Center, Sprague-Dawley rats (aged P24) were subjected to severe CCI injury using previously established methods (Brody et al., 2007; Dixon et al., 1991). Briefly, the rats were deeply anesthetized with isoflurane (3–5%, inhalation to effect) in oxygen (0.8 L/min), and then fixed in a stereotaxic frame. The heads of the rats were shaved and swabbed with povidone-iodine, and the skull was then exposed by a midline scalp incision. A 6-mm craniotomy centered between the lambda and the bregma was made over the right somatosensory cortex. Care was taken not to damage the dura. Severe cortical contusion was produced using an electromagnetic impactor (myNeurolab, St. Louis, MO) with a 5-mm-diameter tip that was driven to a depth of 2.0 mm at a velocity of 4 m/sec. Immediately after injury, isoflurane was decreased to 1.2–2.0%, and a plastic skull cap was secured over the impact site with veterinary adhesive. The skin was then sutured closed and the area was swabbed with antibiotic ointment. The rats were fully ambulatory within 20–40 min after surgery. No animal mortalities resulted from this procedure. Sham-injured control animals were subjected to anesthesia, craniotomy, and recovery, but not CCI. All rats were continuously monitored in their cages for > 6 h following surgery and then returned to the animal room, where they were monitored twice daily. Spontaneous generalized convulsive seizures were observed in one rat immediately after CCI, but these subsided quickly and were not observed thereafter.
Stimulation and statistical analysis
To assess the development of injury-induced changes in cortical neural circuit excitation, electrophysiological recordings were made from acute neocortical brain slices prepared from CCI-injured rats at 7–9 days post-injury (week 1, n = 3 rats), and at 14–16 days post-injury (week 2, n = 6 rats). Using methods previously described (Ling and Benardo 1995; Yang and Benardo 1997), 400-μm-thick coronal slices of somatosensory cortex were prepared from CCI-injured rats (4 slices per rat), from cortical regions ipsilateral and directly adjacent to the injury site (1.5–2.0 mm rostral and/or caudal), or from the corresponding regions in sham-injured control rats. In brief, the rats were deeply anesthetized via inhalational halothane and then decapitated. Their brains were rapidly removed and placed in ice-chilled saline. The somatosensory cortex was isolated and then sectioned into coronal slices using a Vibratome tissue sectioner (Vibratome, Co., St. Louis, MO). The slices were transferred to an interface recording chamber (Fine Science Tools, Foster City, CA) and maintained at 30 ± 1°C in a humidified oxygenated atmosphere (95% oxygen + 5% CO2). The slices were continuously perfused (∼ 1 mL/min) with artificial cerebrospinal fluid, which was composed of 124 mN NaCl, 5 mM KCl, 26 mM NaHCO3, 1.6 mM MgCl2, 2 mM CaCl2, and 10 mM glucose, with a pH of 7.35–7.40 when saturated with 95% oxygen and 5% CO2 (Hoffman et al., 1994). All slices were allowed to equilibrate in the recording chamber for 1–2 h prior to recording. Standard intracellular and extracellular techniques were used to record neuronal activity in neocortical layer V (Yang and Benardo, 1997). Membrane potentials were measured using an AxoClamp 2B amplifier operating in current-clamp mode (Axon Instruments, Foster City, CA). For intracellular recordings of individual neurons, glass microelectrodes with tip resistances of 60–80 MΩ were filled with 2 M potassium acetate. For extracellular recordings of field potentials, electrodes were filled with 1 M NaCl and had tip resistances of 2–5 MΩ. All signals were recorded directly to computer hard disk using pCLAMP software (Axon Instruments). Neural responses were evoked by stimulating slices with cathodal shocks (40–200 μA, 200-μsec duration), delivered at low frequency (0.067 Hz), via bipolar tungsten electrodes placed at the border of layer VI and white matter. Following all recordings, the slices were fixed (4% paraformaldehyde in 0.1 M phosphate buffer; pH 7.3) for histological analyses. Slices that were not resectioned were stained with Fluoro-Jade B (FJB) to assess injury-induced cell necrosis (Schmued and Hopkins, 2000). FJB-stained slices were then mounted and viewed under confocal epifluorescence (Zeiss LSM 510 with Axioscope 2; Carl Zeiss, San Diego, CA) with a blue spectrum light source (450–490 nm) and barrier notch filter for fluorescein (515–565 nm). Slices were scored for neuronal degeneration via visual counts of FJB-positively stained cells, which were made from four randomly selected fields (450 μm × 450 μm) in each slice. To visually assess the extent of the CCI-induced lesion, separate coronal slices were made at the cortical injury site and immediately fixed (i.e., not used for electrophysiological recordings), and then resectioned (50 μm) for Nissl staining. All data are expressed as means ± SEM. Statistical significance was determined using Student's t-test at the 0.05 level.
Results
Histological examination of brain slices showed that CCI injury produced severe focal damage in the ipsilateral somatosensory cortex. By 2 weeks after injury, the gray matter lost its normal conformation at the concussion site, and further examination showed that overall cortical damage extended beyond the dimensions of the 5-mm impactor tip (Fig. 1A). A cavity formed with irregular edges that extended to the hippocampal structure. Analysis of Nissl and FJB staining indicated that there was significant cell necrosis at both 1 and 2 weeks after injury (Fig. 1B and C), suggesting an ongoing process of active neuronal degeneration in neocortex. No laminar-specific pattern of cortical cell degeneration was observed.
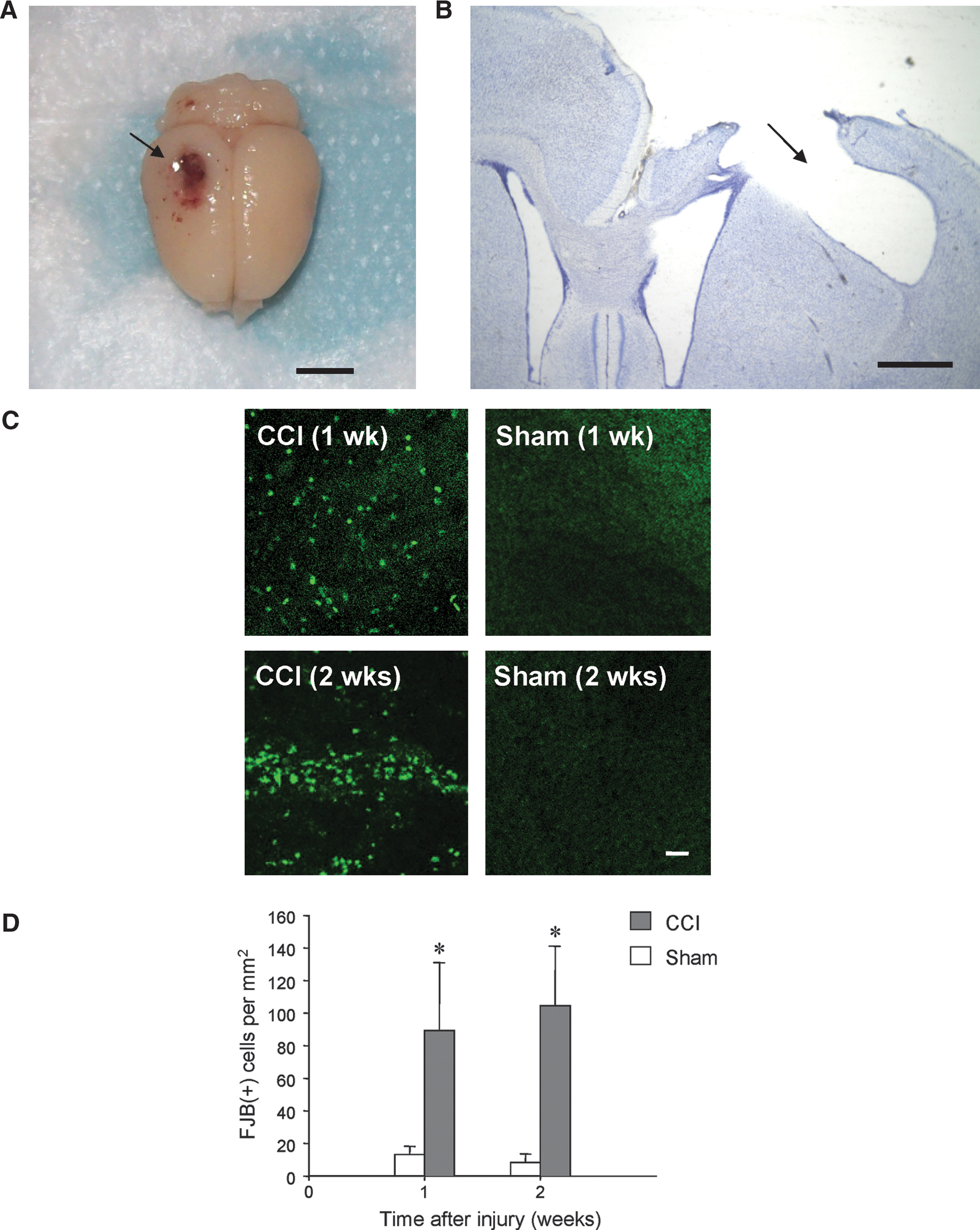
Neuronal degeneration in rat neocortex after CCI injury. (
To assess the development of post-traumatic epileptiform activities, electrophysiological recordings were made from neocortical layer V in slices prepared at 1 and 2 weeks after injury. All intracellular recordings were obtained from neurons physiologically identified as regular-spiking pyramidal cells, with a mean resting potential (RMP) of −71 ± 1.5 mV, and input resistance of 32.5 ± 1.6 MΩ (n = 25). We examined 11 slices from 3 rats at 7–9 days post-injury (week 1), and 24 slices from 6 rats at 14–16 days post-injury (week 2). As a control, 16 slices from 4 sham-injured rats were also examined at these time points. Hyperexcitability and the expression of epileptiform discharges were determined using two measures: (1) the duration of burst discharges of action potentials (APs) evoked by externally-applied stimuli, and (2) the presence or absence of spontaneous epileptiform burst activity. Burst duration was measured as the time from the peak of the first AP to the time the last AP decayed to 10% of its peak amplitude, and was taken from intracellular recordings that were closely synchronized with population potentials recorded extracellularly. Burst discharges with durations > 2 sec were classified as ictal-like (D'Antuono et al., 2010; Köhling et al., 2000; Traub et al., 1996), since the occurrence of these synchronized events in vivo would likely be classified clinically as electrographic seizures (Fuortes et al., 2008). Shorter-duration bursts (≤2 sec) were classified as interictal-like events. Both evoked and spontaneous epileptiform activity that included interictal-like and ictal-like firing were observed in neocortical layer V in slices prepared over the course of the 2-week period following CCI injury. This was in contrast to the activity recorded in slices from sham-injured animals (Fig. 2A), in which application of graded intensity stimuli evoked excitatory postsynaptic potentials (EPSPs) of increasing magnitude ultimately triggered single AP discharges, even at supra-threshold stimulus intensities.
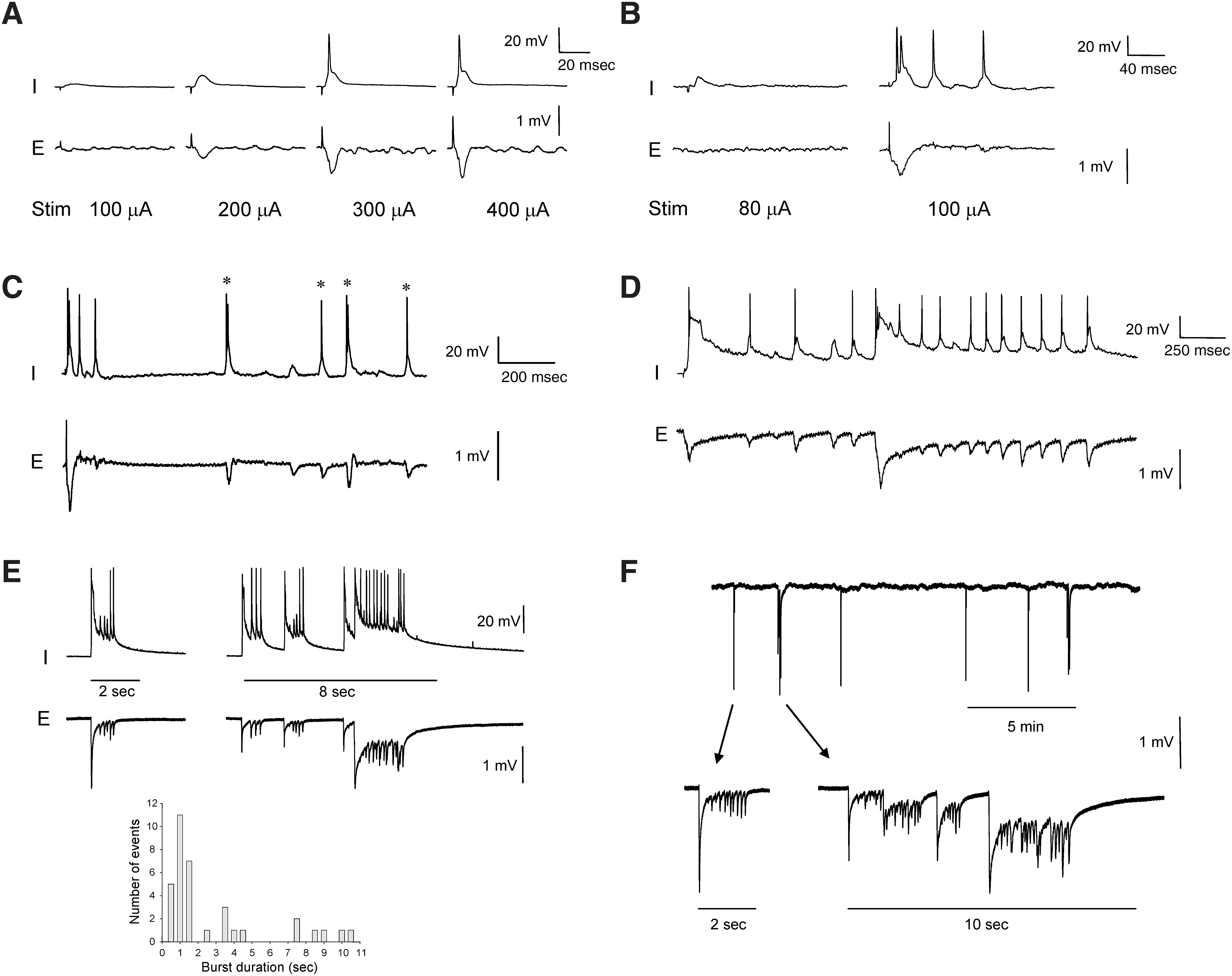
Epileptiform burst discharges in rat neocortex after CCI injury. (
In slices prepared from CCI-injured rats at week 1, intracellular recordings from pyramidal cells revealed evoked epileptiform events that were synchronized with population events recorded extracellularly. These events were triggered at threshold stimulus intensity in an all-or-none manner, and were characterized by bursts of 3–5 APs (mean 3.4 ± 0.3 APs) superimposed on a paroxysmal depolarization shift (Fig. 2B). The average burst duration was brief (<1 s), with a mean value of 174.0 ± 84.2 msec (n = 8 slices; range 39.5–747.9 msec), suggestive of interictal-like firing. The primary bursts were followed by recurrent, repetitive after-discharges of varying latency (Fig. 2C). At week 1, over 80% of slices (9 of 11) from CCI-injured animals were hyperexcitable, compared to none (0 of 8) of slices from sham-injured rats.
In slices prepared from CCI-injured rats at week 2, recordings revealed both spontaneous and evoked epileptiform events. Similarly to week 1, external stimuli triggered epileptiform bursts in > 80% of slices (20 of 24) in an all-or-none manner. However, evoked bursts were significantly longer (by ∼ 7-fold), with a mean duration of 1281.8 ± 273.4 msec (range 330.1–3020.5 msec; p < 0.05 relative to week 1), and 4–24 APs per burst (mean 9.9 ± 1.9 APs; p < 0.05). Most notably, spontaneous epileptiform discharges were observed in slices (n = 3) from 2 of 6 CCI-injured rats (33%). Spontaneous activity consisted of both brief (≤2 sec) interictal-like bursts, and prolonged (up to 8–10 sec) ictal-like discharges (Fig. 2D and E), with ictal-like events representing 37.0% of spontaneous burst discharges. A frequency histogram of spontaneous burst duration values revealed a skewed distribution, with a modal value of 1 sec and smaller peaks at 3.5 and 7.5 sec (Fig. 2E bottom panel; n = 35 events, bin size = 0.5 sec). The frequency of spontaneous bursts varied between the two rats, with a mean inter-burst interval of 78.6 ± 19.4 sec (range 4.5–289.1 sec) in one slice from one rat, and 206.6 ± 49.7 sec (range 108.5–419.2 sec) in two slices from the second rat (Fig. 2F). These results indicate a progressive development of hyperexcitability in neocortical circuits during the first 2 weeks following severe CCI injury, as shown by the significant increase in duration of evoked epileptiform events and the emergence of spontaneous epileptiform activity over this period. Together, these observations suggest that a rapid epileptogenic process occurs after severe cortical TBI.
Discussion
In this study we examined the changes in neural circuit excitability seen in rats following CCI injury, and report for the first time the development of spontaneous ictal-like discharges in rat neocortex after cortical contusion. Previous studies have examined the cellular and molecular mechanisms underlying post-traumatic epileptogenesis using other chronic injury models or rodent species, including lateral FPI (D'Ambrosio et al., 2004; Pitkänen et al., 2009), cortical undercutting (Hoffman et al., 1994; Prince and Tseng, 1993; Prince et al., 2009), and CCI in mice (Hunt et al., 2009). These studies have shown the development of spontaneous, post-traumatic behavioral seizures in both rats and mice, that in some cases were paralleled by the development of hyperexcitability assessed in vitro in acute brain slices prepared from experimentally injured animals. However, though these studies show the development of behavioral seizures, increased seizure susceptibility, and both evoked and spontaneous epileptiform events in brain slices, none reported spontaneous ictal-like discharges from neocortex.
CCI produces a reproducible, focal gray matter injury that differentiates it from other TBI models, including FPI and undercut injury. CCI gives rise to a spectrum of physiological responses and anatomic damage that resemble some aspects of closed head injury seen clinically. Lateral CCI injury is an established brain trauma model that was originally thought to induce only a focal insult, but recent studies suggest more widespread degeneration in white matter, hippocampus, and thalamus (Hall et al., 2008). Nonetheless, CCI produces a more focused injury than that of FPI, and has the advantage of precisely controlled injury variables such as deformation depth and impact velocity, which form highly reproducible contusion cavities in the neocortex. In this study, an impact 2 mm in depth caused substantial gray matter damage, resulting in cavity formation similar to that reported for severe TBI in rodents (Goodman et al., 1994) and mice (Hunt et al., 2009). We found that at 1 week post-injury, evoked epileptiform burst firing could be recorded in neocortical slices prepared from sites directly adjacent to the injury focus. Over the course of the first 2 weeks following injury, neocortical circuits became progressively more hyperexcitable, as manifested as prolonged evoked burst discharges and spontaneous epileptiform activities that included both ictal-like and interictal-like discharges. The hallmark of PTE is chronic and recurrent unprovoked seizures. Late or chronic seizures are classically defined as episodes that occur more than 1 week following neurotrauma, and are considered positive predictors of PTE (Frey, 2003). In humans, the latent period between the injury event and the onset of late seizures may extend from weeks to years, though the epileptogenic process likely begins immediately after TBI (Yang et al., 2007). The initial insult triggers a cascade of cellular and molecular events, that over the course of the latent period bias cortical networks towards hyperexcitation, until the threshold is reached for recurrent spontaneous seizures. Studies using the undercut cortex model suggest that injury-induced hyperexcitability and epileptiform activity may arise from several mechanisms that are triggered in response to the depression of activity that results from neuron loss, including increased axonal sprouting and homeostatic synaptic plasticity (Hoffman et al., 1994; Prince and Tseng, 1993; Timofeev et al., 2010). Our previous studies of superficial cortical injury suggest that trauma-induced increases in glutamate receptor conductance may also play a role, particularly in the acute phase immediately following injury (Yang et al., 2007). However, it is not yet known whether similar epileptogenic mechanisms are involved in CCI. Moreover, it remains to be seen whether the progression in neocortical hyperexcitability observed after CCI in rats leads directly to behavioral seizures or PTE. It will be it necessary to perform in vivo electroencephalogram (EEG) or electrocorticogram (ECoG) recordings, as well as long-term video monitoring of animals for behavioral seizure activity, to determine whether CCI-injured rats develop epilepsy (Hudak et al., 2004).
From the data presented in this preliminary study, we do not yet know whether the epileptiform activities are limited to the injury focus or spread to a wider brain area. Chronic in vivo EEG or ECoG recordings from rats may be useful in clarifying this issue (Timofeev et al., 2010). As shown by other studies of cortical injury (D'Ambrosio et al., 2004; Hunt et al., 2009; Prince and Tseng, 1993), in vitro slice recordings may also be useful as complements to in vivo recordings in assessing the development of early or latent hyperexcitability in local cortical networks, as well as changes in neural circuit properties, since stable paired intracellular and extracellular recordings are easily obtained in slices. In addition, the in vitro environment can be easily manipulated to uncover latent hyperexcitable states via electrical and chemical stimulation. Both evoked and spontaneous epileptiform discharges have been observed in neocortical slices prepared from rats 2–3 weeks after undercut injury, in the absence of pro-convulsant drugs (Hoffman et al., 1994; Prince and Tseng, 1993). However, these events were limited to brief interictal-like events, with no prolonged ictal-like discharges observed. Evoked ictal-like discharges lasting up to 10 sec were observed in rare instances in slices from guinea pigs, but no spontaneous ictal-like activity was observed (Hoffman et al., 1994). The absence of spontaneous ictal-like discharges (as compared to those seen after CCI) may stem from inherent differences in the type of injury, and possibly even with the animal species used, since evoked ictal discharges were not seen in rats with undercut lesions. In the undercut model, the initial primary insult involves more white matter than gray matter damage (Hoffman et al., 1994), which may require a longer latency period for development of spontaneous epileptiform events (particularly ictal activity) than the extensive gray matter injury directly caused by CCI. Studies using the lateral FPI model have reported chronic electrographic cortical seizures in rats at 8–10 weeks following injury (D'Ambrosio et al 2004), and behavioral seizures at 7 weeks to 1 year post-injury (Kharatishvili et al., 2006; Pitkänen et al., 2009). Chronic behavioral seizures have also been observed in CCI-injured mice at 6–10 weeks after trauma (Hunt et al., 2009). However, none of the FPI or CCI studies recorded spontaneous epileptiform firing in acute brain slices prepared from injured animals, though evoked burst discharges were seen in acute slices of rat neocortex after FPI (D'Ambrosio et al., 2004), and mouse hippocampus following CCI (Hunt et al., 2009). Hunt and associates (2009) reported spontaneous epileptiform firing in hippocampal slices from CCI-injured mice in the presence of convulsant media composed of Mg2+-free saline and picrotoxin (100 μM), but not in normal artificial cerebrospinal fluid. It is unclear whether the absence of spontaneous seizure-like activity in these other studies reflects differences in rodent species, injury modalities, or neocortical versus hippocampal responses to trauma. The extracellular potassium concentration (5 mM) used in this study was slightly higher than those measured in vivo (Somjen, 2002), in order to match other neurotrauma models (Hoffman et al., 1994; Prince and Tseng, 1993), and to help unmask neural circuit hyperexcitation by compensating in part for the decreased levels of network activity in brain slices that result from the deletion of synaptic inputs by the slicing procedure (Destexhe and Paré, 1999; Destexhe et al., 2003). Although this potassium concentration might favor states of increased excitation, it alone is insufficient to elicit spontaneous epileptiform activity, as we found no evidence of increased network excitability, spontaneous synchronized activity, or depolarized pyramidal cell RMPs in slices from sham-injured control animals (cf. Hoffman et al., 1994; Prince and Tseng, 1993). Injury-induced hyperexcitation of neocortical circuits after CCI may stem in part from a decrease in GABAergic inhibition (Yang and Benardo, 1997; Yang et al., 2007), possibly due to a preferential loss of inhibitory interneurons. A recent study using the cortical undercut model showed that GABAergic neurons may be particularly vulnerable to neurotrauma, with significant declines in both GABA- and GAD-labeled neurons within 2–6 weeks after injury (Avramescu et al., 2009). We observed active neuron degeneration during the first 2 weeks after CCI that paralleled the development of hyperexcitation and epileptiform activities, though we do not yet know which specific subpopulations of cortical neurons are affected in this model. However, a selective loss of inhibitory cells following CCI would upset the delicate balance between synaptic excitation and inhibition in the neocortex (Ling and Benardo, 1995), leading to a lower seizure threshold and increased seizure susceptibility (Yang et al., 2007). Further histological analyses are needed to address these issues.
To our knowledge, this investigation represents the first study to demonstrate chronic development of spontaneous ictal-like epileptiform discharges in the neocortex following cortical contusion injury in rats. Although we did not systematically monitor behavioral seizures in these animals, our findings suggest a progressive lowering of the seizure threshold within the first 2 weeks after injury. Though the neocortex appears to have a higher threshold for epileptogenesis than other brain regions (Abdelmalik et al., 2005; Racine 1975), past studies suggest that direct gray matter lesions lead to epileptogenic activity in the neocortex first, which may then progress to other areas (D'Ambrosio et al., 2005). Our findings indicate that hyperexcitation develops rapidly in neocortical circuits after direct cortical injury, leading to evoked and spontaneous seizure-like discharges that may reflect the early stages of post-traumatic neocortical epileptogenesis. Future experiments will further assess the temporal and spatial patterns of CCI-induced epileptogenesis to determine whether these lead to more widespread expression of epileptiform activity throughout the cortex, and a shift in spontaneous activity towards more prolonged, ictal-like events. Thus CCI could be a valuable model for elucidating the mechanisms underlying neocortical PTE and help identify potential targets for prophylactic interventions.
Footnotes
Acknowledgments
The authors wish to thank Dr. Robert K.S. Wong for helpful comments and suggestions in the preparation of this manuscript, and Dr. David Brody and Katherine Schwetye for generously providing training and instruction in the CCI technique. This work was supported by funds from the Research Foundation of SUNY Principal Investigator Incentive program (to D.S.F.L).
Author Disclosure Statement
No competing financial interests exist for any of the authors of this manuscript.