
Abstract
We have reported differential short- and long-term dysregulation of the neuroendocrine stress response after traumatic brain injury (TBI) produced by controlled cortical impact (CCI). We have now investigated three possible mechanisms for this TBI-induced dysregulation: (1) effects on the sensitivity of negative-feedback systems to glucocorticoids; (2) effects on the sensitivity of pituitary corticotrophs to corticotropin-releasing hormone (CRH); and (3) effects on neuronal loss in the hilar region of the dentate gyrus and in the CA3b layer of the dorsal hippocampus. TBI was induced to the left parietal cortex in adult male rats with a pneumatic piston, at two different impact velocities and compression depths, to produce either moderate or mild CCI. At 7 and 35 days after surgery, the rats were injected SC with the synthetic glucocorticoid analog dexamethasone (DEX; 0.01, 0.10, or 1.00 mg/kg) or saline, and 2 h later were subjected to 30 min of restraint stress and tail vein blood collection. Whereas all doses of DEX suppressed corticosterone (CORT) and adrenocorticotropic hormone (ACTH) responses to stress on both days, CORT and ACTH were significantly more suppressed after 0.01 mg/kg DEX in the moderate TBI group than in the mild TBI or sham groups. At both 7 and 35 days post-TBI, CRH (1.0 and 10.0 μg/kg IP) stimulated CORT and ACTH in all rats, regardless of injury condition. Hippocampal cell loss was greatest at 48 days after moderate TBI. Enhanced sensitivity to glucocorticoid negative feedback and greater hippocampal cell loss, but not altered pituitary responses to CRH, contribute to the short- and long-term attenuation of the neuroendocrine stress response following moderate TBI. The role of TBI-induced alterations in glucocorticoid receptors in limbic system sites in enhanced glucocorticoid feedback sensitivity requires further investigation.
Introduction
Studies with experimental models of TBI have demonstrated acute activation of the hypothalamic-pituitary-adrenal (HPA) axis after injury. For example, mRNA for corticotropin-releasing hormone (CRH) is upregulated at 2 and 4 h after fluid percussion injury (FPI; Grundy et al., 2001; Roe et al., 1998), and adrenocorticotropic hormone (ACTH) and corticosterone (CORT) are increased within the first 6 h after controlled cortical impact (CCI) injury (Gottesfeld et al., 2002; McCullers et al., 2002a; Taylor et al., 2004), and within 8 h after closed head injury (Shohami et al., 1995). A recent study reported long-term effects of medial frontal cortex CCI on serum growth hormone, interleukin-1β, and glial fibrillary acidic protein in the hypothalamus and anterior pituitary (Kasturi and Stein, 2009). We previously reported that TBI produced by moderate lateral CCI causes long-term dysregulation of the neuroendocrine stress response (Taylor et al., 2006). In a subsequent study (Taylor et al., 2008), we continued to observe attenuation of the HPA stress response for as long as 70 days after moderate CCI, while a milder CCI injury facilitated the stress response.
Neural and hormonal mechanisms play important roles in the regulation of the HPA axis. To further study the effect of TBI on HPA regulation, in this study we evaluated the potential role of two hormonal and one neural mechanism in mediating the facilitated and/or attenuated HPA responses to an acute, novel stressor in rats with mild or moderate CCI. First, in order to examine the effects of TBI on the sensitivity of negative-feedback systems to glucocorticoids, ACTH and CORT responses to restraint stress were determined following peripheral administration of the synthetic glucocorticoid analog dexamethasone (DEX). Second, to examine the effects of TBI on the sensitivity of pituitary corticotrophs to CRH, ACTH and CORT responses were determined following peripheral CRH administration. Finally, having previously reported (Taylor et al., 2008) that TBI severity differentially affected cell loss in the hippocampus, a brain region that primarily inhibits the HPA axis, in this study we examined the long-term effects of TBI on subfields of the hippocampus in greater detail. A preliminary report has previously been published (Taylor et al., 2009).
Methods
Subjects
Adult male Sprague-Dawley rats (340 ± 6.69 g) from Charles River Breeding Labs (Hollister, CA) were used as experimental subjects (n = 144). Upon arrival, the rats were acclimated for 1–2 weeks to our standard temperature and lighting conditions (22 ± 1°C, 14-h/10-h lighting cycle, lights on 0400–1800 h). All experimental procedures were approved by the UCLA and VAGLAHS Institutional Animal Care and Use Committees.
Surgical procedures
At 60–70 days of age, the animals were anesthetized with isoflurane (2.0–2.5% in 100% oxygen, 2.0-mL/min flow rate) and placed into a stereotaxic frame (Kopf Instruments, Tujunga, CA), with the head positioned in a horizontal plane with respect to the interaural line. During all surgical procedures, body temperature was maintained at 37–38°C using a thermostatically controlled heating pad (Harvard Apparatus, Holliston, MA). All surgical procedures were performed under aseptic conditions and have been previously described in detail (Sutton et al., 1993; Taylor et al., 2002, 2006, 2008). In brief, following a midline incision, the skin, fascia, and temporal muscle were reflected. Animals receiving CCI were subjected to a 6-mm-diameter craniotomy over the left parietal cortex centered at 3 mm posterior and 4 mm lateral to the bregma. An electronically-controlled, small-bore, dual-stroke pneumatic piston cylinder with a 40-mm stroke (Hydraulics Control, Inc., Emeryville, CA) was mounted onto a stereotaxic micromanipulator (Trentwells Co., Coultervillle, CA), allowing for precise control of the impact site and depth of tissue compression. The piston cylinder was angled 22° from vertical, enabling the flat, circular impactor tip (5 mm diameter) to be perpendicular to the surface of the brain at the site of injury. Severity of injury was adjusted by altering the velocity used to impact the exposed dura, and by altering the depth of tissue compression (moderate CCI: 30 psi or ∼2.8 m/sec, 2.0 mm; mild CCI: 20 psi or ∼2.2 m/sec, 1.5 mm; Dixon et al., 1991; Sutton et al., 1993). Following induction of the moderate or mild CCI, the fascia and scalp were sutured closed, and the wound margins were infiltrated with local anesthetic (1% xylocaine). Sham-operated control rats underwent similar procedures to control for surgical stress and duration of anesthesia, but did not receive craniotomy or any impact. The animals were returned to their cages after recovery from anesthesia, where they were pair-housed and subsequently subjected to the experimental procedures outlined below.
Dexamethasone treatments and restraint stress
Dexamethasone (DEX; Baxter Healthcare Corp., Deerfield, IL) was purchased in 10-mg/mL vials and diluted with sterile 0.9% saline to the desired concentrations. At 7 and 35 days post-surgery, after basal blood collection by tail venipuncture, the rats received an injection of DEX (0.01, 0.10, or 1.00 mg/kg SC) or saline. Two hours following DEX/saline administration, restraint stress was initiated. The animals were placed into flat-bottom acrylic glass restraining tubes (13 × 6 × 8 cm; Harvard Apparatus) and tightly restrained for 30 min, followed by light restraint for an additional 60 min. On both drug treatment days, additional blood samples were obtained by tail venipuncture at 30, 60, and 90 min after stress onset. All testing occurred between 0900 and 1300 h. The experiment was replicated four times, once for each dose of DEX/saline, with each replicate consisting of 4–10 mild-, moderate-, and sham-injured rats per group.
Corticotropin-releasing hormone treatments
Corticotropin-releasing hormone (CRH human, rat; Sigma-Aldrich, St. Louis, MO) was purchased in vials of 1 mg/mL water and diluted with sterile 0.9% saline to the desired concentrations. At 7 and 35 days post-surgery, after basal blood collection by tail venipuncture, the rats received an injection of CRH (1.0 or 10.0 μg/kg IP) or saline. At 30, 60, 90, and 240 min after injection of CRH, with the rats lightly restrained, blood was collected by tail venipuncture. Plasma was processed for CORT and ACTH analysis as described below. Each dose of CRH or saline was injected into six moderate CCI and sham injured rats that had not been used for the DEX studies.
Assay of plasma content of corticosterone and adrenocorticotropic hormone
Blood samples (300 μL) were collected at the time-points described above, in microcapillary blood collection tubes that contained EDTA with added aprotonin (200 kIU/mL). After centrifugation at 2000 RPM for 20 min, plasma was separated and stored at −80°C until analysis for CORT and ACTH.
Plasma CORT was assessed with a rat CORT 125I RIA kit (MP Biomedicals, Irvine, CA), as we have done previously (Taylor et al., 2002). The reported detection limit of this assay is 8 ng/mL, the recovery of exogenous CORT is 100%, and intra- and inter-assay coefficients of variation are lower than 10.3% and 7.2%, respectively. The results are expressed as nanograms per milliliter of plasma.
Plasma ACTH levels were assessed with a human ACTH 125I RIA kit (DiaSorin, Stillwater, MN) specific for ACTH1-39, as done previously (Taylor et al., 2002). The reported detection limit of this assay is 15 pg/mL, the recovery of exogenous ACTH is greater than 87.5%, and intra- and inter-assay coefficients of variation are lower than 10.7% and 5.7%, respectively. The results are expressed as picograms per milliliter of plasma.
Histology
The rats were sacrificed by decapitation at 48 days after surgery without any further exposure to DEX. The scalp and temporal muscles were retracted from the skull surface and the head was submerged in 10% formalin for ∼3 weeks. Brain tissue was removed from the calvarium and placed in fresh 10% formalin for 1 week. Brains from five randomly selected rats in each of the experimental DEX and saline groups (20 shams, 20 mild TBI, and 20 moderate TBI) were transferred to a solution of 20% sucrose in 10% formalin. After sinking in the sucrose/formalin solution, the brains were frozen and sectioned in the coronal plane (25 μm thick), with every tenth section being saved and stained with thionine.
Microscopic analyses of hippocampal damage were performed using bright-field illumination with a Leica microscope that was interfaced with a computer and Stereo Investigator software (v.3.0; MicroBrightField Inc., Colchester, VT). Surviving polymorphic neurons within the hilar region of the dentate gyrus, and pyramidal neurons within the CA3b layer of the dorsal hippocampus were counted on three tissue sections (250 μm between sections) lying between −2.8 and −3.8 mm from the bregma (Paxinos and Watson, 2005). Two-dimensional contours encompassing the polymorph layer of the dentate gyrus (PoDG) lying between the upper and lower blades of the dentate granule cells, but excluding the CA3c/CA4 pyramidal cell layer, were placed bilaterally on each tissue section. Two rectangular box contours (200 × 100 μm; W × H) were placed bilaterally over the cells in the CA3b pyramidal layer; one box was positioned over medial CA3b neurons lying 200–400 μm lateral to a line connecting the lateral tips of the upper and lower blades of the granule cells, and the other box was positioned over lateral CA3b neurons lying 200–400 μm medial to the lateral edge of the CA3b layer. Cell density counts were made for all neurons lying within each of the left and right contour regions by an experimenter blinded to drug treatment condition. The counts from PoDG, lateral CA3b, and medial CA3b regions were summed across the three tissue sections for the left and right hemispheres for each animal, and expressed as numbers of cells per square millimeter prior to calculating group averages.
For six randomly selected rats sustaining sham, mild, or moderate injury, the area of the cortical mantle on every other thionine-stained section (19 sections, 500 μm apart) between AP + 1.7 and −7.3 from the bregma (Paxinos and Watson, 2005) was viewed, traced, and quantified using computer-assisted morphometry (SCION/NIH Image 4.0.3.2; National Institutes of Health, Bethesda, MD), and the left and right cortical tissue volume was calculated as previously described (Sutton et al., 1993). The percentage of tissue loss at each AP plane, as well as for the entire left/injured cortical mantle between AP + 1.7 and −7.3 was calculated using the formula [100 − ([left/right] × 100)], as previously described (Fukushima et al., 2009; Taylor et al., 2008).
Statistical analysis
Analyses of variance (ANOVA) were carried out on the data, as indicated. Individual group means were compared using t-tests under the Tukey-Fisher least significant difference criterion, with α set at 0.05 (two-sided).
Results
Effect of dexamethasone on corticosterone responses to restraint after mild or moderate TBI
Two hours prior to restraint, the rats were injected with saline or DEX (0.01, 0.10, or 1.00 mg/kg) at 7 and 35 days after mild or moderate TBI or sham injury. Figure 1 shows plasma CORT responses prior to DEX/saline injection, and at 30, 60, and 90 min after restraint on day 35. Comparable data for day 7 are not shown, since the results were virtually the same except for less overall feedback suppression of CORT levels with the lowest dose of DEX.

Effects of prior injection of saline or dexamethasone (DEX) at 0.01, 0.10, or 1.00 mg/kg on plasma corticosterone (CORT) levels immediately preceding injection (0 min), and at 30, 60, and 90 min after restraint, commencing at 2 h post-injection on day 35 after mild or moderate traumatic brain injury (TBI) or sham injury. The results are means ± standard error of the mean of 8–9 animals per group per dose. In each panel, similar letters denote significant (p ≤ 0.01) differences between injury groups post-TBI.
Two-way ANOVAs at each time point indicated significant effects of injury [F (2,91) = 4.01, 7.89, and 4.45 (p ≤ 0.02), at 30, 60, and 90 min after restraint onset, respectively], and of DEX dose/saline injection [F (3,91) = 19.33, 111.66, and 139.39 (p < 0.0001) after restraint onset, respectively). A significant interaction of injury and DEX dose/saline occurred at 30 min after restraint onset [F (6,91) = 2.40; p = 0.03], but not at the other time points.
As can be seen in Figure 1, after injection of saline, the CORT response to restraint was significantly (p ≤ 0.01) lower in the moderate TBI group than in either the mild TBI or the sham-injury group. Whereas all three doses of DEX suppressed CORT responses to restraint stress compared to the CORT response of the saline-injected groups, CORT levels of the moderate TBI group were significantly (p = 0.01) lower than those of the mild TBI or sham-injury groups at 30 and 60 min after 0.01 mg/kg DEX, and compared to the mild TBI group at 30 min after 0.10 mg/kg DEX.
Effect of dexamethasone on adrenocorticotropic hormone responses to restraint after mild or moderate TBI
Figure 2 shows plasma ACTH responses prior to DEX/saline injection, and at 30, 60, and 90 min after restraint on day 35. As with the CORT response, comparable ACTH data for day 7 are not shown, since the results were virtually the same except for dose-dependent suppression of ACTH levels at 0.01 and 0.10 mg/kg DEX, in contrast to similar suppression by all three DEX doses on day 35.
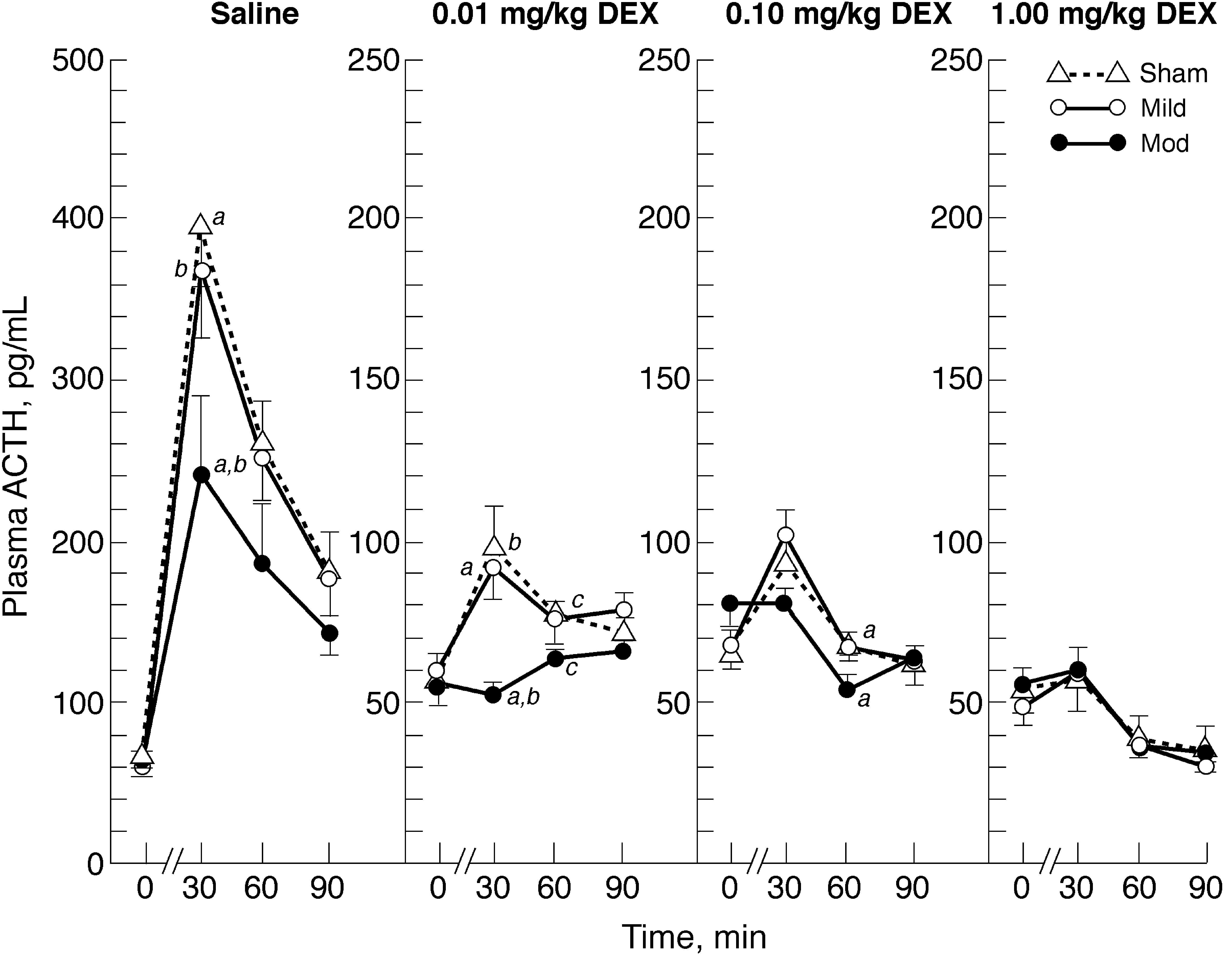
Effects of prior injection of saline or dexamethasone (DEX) at 0.01, 0.10, or 1.00 mg/kg on plasma adrenocorticotropic hormone (ACTH) levels immediately preceding injection (0 min), and at 30, 60, and 90 min after restraint commencing 2 h post-injection on day 35 after mild or moderate traumatic brain injury (TBI) or sham injury. Results are means ± standard error of the mean of 8–9 animals per group per dose. In each panel, similar letters denote significant (p ≤ 0.05) differences between injury groups post-TBI.
Two-way ANOVAs at each time point indicated significant effects of DEX dose/saline injection [F (3,95) = 4.82, 88.41, 89.33, and 75.89 (p < 0.01) at each time point before and after restraint onset, respectively), and a significant effect of injury at 30 min after restraint onset [F (2,95) = 5.39; p = 0.006]. A significant interaction of DEX dose/saline and injury occurred at 30 min after restraint onset [F (6,95) = 2.52; p < 0.03].
Figure 2 shows that all three doses of DEX suppressed ACTH responses to restraint stress compared to the ACTH response of the saline-injected groups. After both saline and 0.01 mg/kg DEX pretreatment, ACTH levels at 30 min after restraint onset were significantly (p ≤ 0.01) lower in the moderate TBI group than in the mild TBI and sham-injured groups. At 60 min after restraint onset, the ACTH levels of the moderate TBI group were significantly lower (p ≤ 0.05) than those of the sham groups that were pretreated with 0.01 or 0.10 mg/kg DEX.
Corticosterone and adrenocorticotropic hormone responses to corticotropin-releasing hormone after mild or moderate TBI
Two-way ANOVA of CORT responses at 35 days post-TBI indicated no significant effects of injury at 0, 30, 60, 90, and 240 min post-injection of 1.0 or 10.0 μg/kg CRH or saline. At 30, 60, 90, and 240 min, there were significant effects of CRH/saline dose [F (2,30) = 8.06, 13.99, and 15.34 (p ≤ 0.001), respectively], but there was no significant interaction of the two factors. At each time point CORT responses were dose-dependent, and the greatest responses occurred at 60 min post-injection. Thus in Table 1 we show data only for 60 and 240 min, and since essentially similar responses occurred at 7 days post-TBI, only the day-35 data are shown.
Results are mean ± standard error of the mean in six animals per group per dose.
Two-way ANOVA of ACTH responses at 35 days post-TBI indicated no significant effects of injury at 0, 30, 60, 90, and 240 min post-injection to 1.0 or 10.0 μg/kg CRH or saline. At 30, 60, and 90 min, there were significant effects of CRH/saline dose [F (2,30) = 33.25, 46.49, and 26.67 (p < 0.0001), respectively], but there was no significant interaction of the two factors. At each time point ACTH responses were dose-dependent, the greatest responses occurred at 30 and 60 min post-injection (Table 1), and hence the greatest CORT responses were seen also at 60 min as described above.
Hippocampal cell loss at 48 days after mild or moderate TBI
Although no persistent effects of prior (day 7 and 35) treatment with DEX were anticipated on day 48, cell density counts in the PoDG region of the dorsal hippocampus were first analyzed using a 3 (injury) × 4 (drug) × 2 (hemisphere) ANOVA. This analysis indicated a significant main effect of injury [F (2,96) = 41.26; p < 0.001], hemisphere [F (1,96) = 187.76; p < 0.001], and the injury × hemisphere interaction [F (2,96) = 37.04; p < 0.001]. Since there was no significant main effect of drug, this data set was further analyzed by collapsing across drug conditions (Fig. 3B). One-way ANOVA of PoDG cell densities revealed no significant effect of injury in the hippocampus contralateral to TBI, while cell densities were significantly altered by injury conditions in the ipsilateral PoDG [F (2,57) = 72.52; p < 0.001]. Post-hoc analyses indicated a significant loss of PoDG neurons ipsilateral to mild (p < 0.001) and moderate (p < 0.001) TBI compared to shams. This cell loss in the ipsilateral PoDG was significantly increased after moderate compared to mild TBI (p < 0.001).

(
Analyses of lateral CA3b cell density counts (Fig. 3C) was performed by collapsing across drug conditions, with a 3 (injury) × 2 (hemisphere) ANOVA revealing a significant main effect of injury [F (2,112) = 23.20; p < 0.001], hemisphere [F (1,112) = 37.90; p < 0.001], and the injury × hemisphere interaction [F(2,112) = 16.08; p < 0.001]. One-way ANOVA of lateral CA3b cell densities revealed no significant effect of injury in the contralateral hippocampus, but similar analyses revealed a significant injury effect in the left/injured hippocampus [F (2,56) = 27.21; p < 0.001]. Post-hoc analyses of this main effect revealed an injury severity–dependent loss of lateral CA3b cells ipsilateral to CCI, with significant neuronal loss occurring after either mild (p < 0.01) or moderate (p < 0.001) TBI compared with sham injury. Cell loss in this lateral CA3 region was significantly greater after moderate than mild TBI (p < 0.001).
Medial CA3b cell density counts (Fig. 3D) were also analyzed using 3 (injury) × 2 (hemisphere) ANOVA, which showed significant main effects of injury [F (2,112) = 4.57; p < 0.05], hemisphere [F (1,112) = 9.69; p < 0.005], and the injury × hemisphere interaction [F (2,112) = 4.65; p < 0.05]. One-way ANOVA revealed no significant effect of injury in the contralateral hippocampus, but a one-way ANOVA for medial CA3b cell densities in the left/injured hippocampus showed a significant effect of injury condition [F (2,56) = 6.47; p < 0.005]. Post-hoc analyses revealed that, although mild TBI cell numbers in this region did not differ from sham levels, moderate TBI led to more significant cell loss in the medial CA3b than mild TBI (p < 0.05) and sham injury (p < 0.005).
Cortical volume loss at 48 days after mild or moderate TBI
A 3 (injury) × 2 (hemisphere) ANOVA of the data for cortical tissue volume revealed a significant main effect of injury [F (2,30) = 38.61; p < 0.001], hemisphere [F (1,30) = 53.86; p < 0.001], and the injury × hemisphere interaction [F (2,30) = 26.05; p < 0.001]. Since a one-way ANOVA of the volume of tissue in the right cortex at 48 days post-surgery revealed no significant effect of injury in this hemisphere contralateral to CCI, we proceeded to express the extent of the cortical injury in each group as the percentage of tissue loss in the left/injured cortical mantle (100 − [(left/right) × 100]). A one-way ANOVA of the latter data revealed a significant effect of injury [F (2,15) = 51.04; p < 0.001]. The percentages of cortical tissue losses after either mild (11.9 ± 4.0; p < 0.005) or moderate (36.5 ± 2.3; p < 0.001) TBI were significant compared to sham injury (–0.9 ± 0.52), and the volume of cortical tissue loss was significantly greater after moderate than after mild TBI (p < 0.001).
Discussion
The current study demonstrates that moderate TBI enhances sensitivity to the synthetic glucocorticoid analog DEX, in the long term (day 35) after injury, as well the short term (day 7; data not shown). The HPA response to an acute psychogenic stressor (i.e., restraint stress) was more suppressed by low-dose (0.01 mg/kg) DEX in the moderate TBI group than in the mild TBI and sham groups. Higher doses of DEX (0.10 and 1.00 mg/kg) suppressed the HPA stress responses of all groups. Increased glucocorticoid negative feedback sensitivity is commensurate with attenuation of the HPA stress response seen after moderate TBI that we reported previously (Taylor et al., 2008), and that was reconfirmed here in the vehicle (saline)-treated rats. Moreover, attenuation of the HPA stress response in the long (and short) term after TBI (at days 35 and 7 post-injury) was not due to impairment of pituitary responsiveness to CRH, and the good correlation between the ACTH and CORT responses implies normal adrenal function. Whereas the observed greater hippocampal cell loss in the moderate TBI group than in the mild TBI or sham-injured groups may be consistent with the attenuated stress response of the moderate TBI group, it does not account for the enhanced glucocorticoid sensitivity seen in response to low-dose DEX.
The HPA axis is activated in response to stressful systemic and/or psychogenic (e.g., restraint stress) stimuli that potentially disrupt homeostasis. It is well established (Herman et al., 2003) that the HPA response to a stressor involves an immediate surge of ACTH release from the pituitary, and the subsequent secretion of corticosteroids from the adrenal cortex that, in turn, generate a shut-off or negative feedback signal. The ACTH surge is initiated by hypophysiotrophic neurons in the medial parvocellular division of the paraventricular hypothalamic nucleus (PVN) that produce CRH, among other ACTH-releasing factors. The medial parvocellular PVN receives synaptic innervation via neurons projecting from various central nervous system (CNS) structures (e.g., the limbic brain regions and brainstem), which evoke rapid activation of the HPA axis. Thus the loss of neurons seen in the cortical and limbic brain regions such as the hippocampus following CCI is consistent with a dysregulated HPA stress response. Whereas the amygdala acts primarily to activate the HPA axis (Matheson et al., 1971), the hippocampus and the medial prefrontal cortex exert inhibitory effects on HPA-axis responses to stress (Herman et al., 1995; Sullivan and Gratton, 1999). Termination of the HPA stress response is mediated by glucocorticoid negative feedback, and glucocorticoid (and mineralocorticoid) receptors are abundantly expressed in the forebrain (i.e., hippocampus, prefrontal cortex, and amygdala; Akana et al., 2001; Diorio et al., 1993; Herman, 1993). Given our finding of enhanced sensitivity to DEX following moderate TBI, our CCI appears to have impacted glucocorticoid receptor–rich regions in these forebrain structures, other than the limited cortical and hippocampal regions that we examined. A search for the affected regions with more specific histological studies is clearly warranted.
Numerous studies have demonstrated that forebrain glucocorticoid receptors are essential for negative feedback regulation of the HPA axis, in particular glucocorticoid feedback inhibition of acute psychogenic stress responses (Furay et al., 2008). The rapid action of glucocorticoids has been shown to be triggered by the activation of membrane-associated receptors and non-genomic signaling mechanisms (de Kloet, 2000; Haller et al., 2008; Tasker et al., 2006). Evidence indicates that glucocorticoids activate divergent G-protein signaling pathways that act in a synapse-specific manner to suppress excitatory synaptic glutamate inputs, and facilitate inhibitory synaptic GABA inputs to magnocellular (Di et al., 2009), as well as parvocellular (Miklos and Kovacs, 2002), PVN neurons. Whereas limbic cortical regions that regulate HPA activity lack direct projections to the PVN, local GABAergic PVN-projecting neurons can be either activated or inhibited by glutamatergic or GABAergic afferent innervation from upstream limbic or cortical regions that are stress responsive and regulate the HPA axis (e.g., ventral subiculum, medial prefrontal cortex, amgydaloid nuclei, and lateral septum; Cullinan et al., 2008).
With respect to TBI, recent reports provide evidence for an association between physical and psychological injuries, such as post-traumatic stress disorder (PTSD) and depression, among other psychiatric disorders (Carlson et al., 2010; Walilko et al., 2009). PTSD, in contrast to depression, is generally thought to be associated with increased glucocorticoid suppression in response to low-dose DEX, reflecting enhanced negative feedback inhibition (de Kloet et al., 2007; Yehuda, 2009). Moreover, enhanced glucocorticoid suppression may also be related to traumatic experiences in the absence of PTSD (de Kloet et al., 2007). Experimentally, it has been shown that glucocorticoid receptor mRNA is suppressed in the ipsilateral dentate gyrus at 24 h after CCI (McCullers et al., 2002b). Evidence also exists for elevated levels of glutamate and GABA in microdialysis samples from the dorsal hippocampus immediately after TBI induced by lateral FPI (Zhong et al., 2006). Other studies have identified enhanced immunobinding of GABA in the dentate gyrus and hippocampal subfield CA1 at 2 and 15 days after FPI in rats (Reeves et al., 1997), and increased levels of GAD67 within the hippocampus at 14 days after CCI (Kobori and Dash, 2006). In the CCI model, GAD67 levels were also increased within the medial prefrontal cortex from 1–28 days post-injury (Kobori and Dash, 2006), returning to control levels by 4 months post-CCI (Hoskison et al., 2009). The present study has shown short- and long-term (days 7 and 35 post-injury) effects of CCI on glucocorticoid feedback sensitivity and suppression of the HPA stress response. Whether the attenuated HPA stress response and its enhanced feedback sensitivity that we observed are mediated by specific TBI-induced changes in glucocorticoid receptors and neurotransmitters in limbic system sites awaits further investigation.
Footnotes
Acknowledgments
We thank Sima Ghavim for her expert assistance with the histological preparations. This research was supported by the Department of Veterans Affairs Medical Research Service, and the UCLA Brain Injury Research Center.
Author Disclosure Statement
No competing financial interests exist.