
Abstract
Functional recovery is markedly restricted following traumatic brain injury (TBI), partly due to myelin-associated inhibitors including Nogo-A, myelin-associated glycoprotein (MAG) and oligodendrocyte myelin glycoprotein (OMgp), that all bind to the Nogo-66 receptor-1 (NgR1). In previous studies, pharmacological neutralization of both Nogo-A and MAG improved outcome following TBI in the rat, and neutralization of NgR1 improved outcome following spinal cord injury and stroke in rodent models. However, the behavioral and histological effects of NgR1 inhibition have not previously been evaluated in TBI. We hypothesized that NgR1 negatively influences behavioral recovery following TBI, and evaluated NgR1−/− mice (NgR1−/− study) and, in a separate study, soluble NgR1 infused intracerebroventricularly immediately post-injury to neutralize NgR1 (sNgR1 study) following TBI in mice using a controlled cortical impact (CCI) injury model. In both studies, motor function, TBI-induced loss of tissue, and hippocampal β-amyloid immunohistochemistry were not altered up to 5 weeks post-injury. Surprisingly, cognitive function (as evaluated with the Morris water maze at 4 weeks post-injury) was significantly impaired both in NgR1−/− mice and in mice treated with soluble NgR1. In the sNgR1 study, we evaluated hippocampal mossy fiber sprouting using the Timm stain and found it to be increased at 5 weeks following TBI. Neutralization of NgR1 significantly increased mossy fiber sprouting in sham-injured animals, but not in brain-injured animals. Our data suggest a complex role for myelin-associated inhibitors in the behavioral recovery process following TBI, and urge caution when inhibiting NgR1 in the early post-injury period.
Introduction
The interaction of Nogo-66 with NgR1 may be pharmacologically blocked by a competitive antagonist, NEP1-40 (GrandPre et al., 2002), inhibiting the interaction between Nogo-66 and NgR1, or a truncated, soluble NgR1 [AA-NgR1(310)ecto-Fc; Fournier et al., 2002], inhibiting the interaction between NgR1 and MAG, OMgp, and Nogo-66. When NgR1(310)ecto-Fc or NEP 1-40 were administered following spinal cord injury (SCI) in both rats and mice, or following cerebral ischemia in rats, consistent findings include enhanced axonal outgrowth and sprouting and accelerated functional recovery (GrandPre et al., 2002; Lee et al., 2004; Li and Strittmatter, 2003; Li et al., 2004; Wang et al., 2006). NgR1 knockout mice (NgR1−/−) have also been evaluated following ischemic brain injury and SCI, showing improved motor outcome (Kim et al., 2004; Lee et al., 2004). Following SCI, regeneration of rubrospinal and raphespinal but not corticospinal pathways has been observed (Kim et al., 2004; Zheng et al., 2005), suggesting that some but not all fiber tracts respond to NgR1 inhibition.
Although TBI is a common cause for long-term suffering and morbidity, pharmacological treatment options proven to improve the clinical outcome are lacking to date and are urgently needed (Marklund et al., 2006a). Here, we sought to enhance behavioral recovery in a mouse model of focal TBI using, in separate studies, pharmacological neutralization and genetic deletion of NgR1. We evaluated the behavioral recovery and histological outcome in NgR1−/− mice (NgR1−/− study), and post-injury neutralization of NgR1 signaling using a soluble dominant negative form of the Nogo receptor (sNgR1 study), in mice subjected to a clinically-relevant model of TBI.
Methods
Study design
All animal procedures described herein were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania or at Yale University (NgR−/− study), and performed in accordance with standards published by the National Research Council (National Research Council, 1996), or approved by the Uppsala County Animal Ethics Committee (sNgR1 study).
NgR−/− study
To evaluate the role of NgR1 in TBI, a mouse strain with selective disruption of the ngr gene was used. Briefly, exon 2 of the mouse NgR1 gene was replaced with a neoR cassette, and chimeric mice were generated and crossed onto a C57BL/6J strain, and back-crossed for another four to six generations, and then inter-crossed to yield homozygous knockouts as previously described (Kim et al., 2004). The mutation in these mice deletes exon II of the NgR1 gene, which encodes the entire mature NgR1 protein, and thus no NgR1 mRNA or NgR1 protein is detectable in these mice (Kim et al., 2004). Gross anatomy and white matter tracts are indistinguishable from wild-type controls, and the levels of Nogo-A and MAG are normal in the brains of adult NgR−/− mice (Kim et al., 2004). Here, adult 4-month-old NgR−/− mice (n = 10, four male, six female), and wild-type (WT; n = 10, four male, six female) littermates, were exposed to controlled cortical impact brain injury (CCI), and six sham-injured C57BL/6 male mice were used as controls (in experiments performed at the Department of Neurosurgery, University of Pennsylvania). No obvious phenotypic differences were observed among the NgR1−/− mice, their age-matched, WT littermate controls, or the sham-injured controls. Due to the limited number of the genetically modified mice available at the time of the NgR −/− study, no sham-injured, age-matched mice for each genotype could be made available. Instead, the cognitive function of 10 male age-matched naïve NgR1−/− mice compared to 10 naïve male WT littermate control animals was evaluated by learning and memory tests in the Morris water maze (MWM), in experiments performed at the Department of Neurology, Yale University, New Haven, Connecticut.
Soluble NgR1 treatment (sNgR1 study)
Pharmacological neutralization of NgR1 was evaluated using soluble NgR1 [AA-NgR1(310)ecto-Fc], an engineered variant of the NgR-ecto-Fc fusion protein reported previously (Li et al., 2004). This protein comprises a 310-amino acid fragment of rat NgR1 fused to a rat IgG1 Fc fragment, in which Cys266 and Cys309 were replaced with alanine residues in order to eliminate heterogenous disulfide bonds. The construct was expressed in Chinese hamster ovary cells, protein was purified, and binding to Nogo66, OMgp, and MAG was verified (Li et al., 2005). This modified protein inhibits the Nogo66-NgR interaction and promotes neurite outgrowth in vitro with similar potency as the unmodified NgR-ecto-Fc (P. Weinreb, F. Qian, M.-Y. Jung, and D. Lee, unpublished observations). Mini-osmotic pumps (pump model 2004; Alzet, Cupertino, CA) filled with NgR1(310)ecto-Fc in phosphate-buffered saline (PBS) or vehicle (Harvey et al., 2009; Wang et al., 2006), were connected to Brain Infusion Kit 3 (Alzet) and primed at 37°C for 48 h. Immediately following CCI or sham injury, the pump was placed into a subcutaneous pocket between the scapulae. The brain cannula was stereotaxically inserted into the contralateral ventricle (1 mm caudal to the bregma, 2 mm lateral to the midline, at a depth of 3 mm), and correct placement was verified in all animals using hematoxylin and eosin–stained brain sections. Vehicle or NgR1(310)ecto-Fc at a concentration of 2.5 μg/μL was infused intracerebroventricularly at 0.25 μL/h for 28 days. The sNgR1 study was performed at the Department of Neurosurgery, Uppsala University, and a total of 40 adult 4-month-old male C57BL/6 mice were included in four groups: CCI injury and sNgR1 treatment (n = 11), CCI injury and vehicle treatment (n = 12), sham injury and sNgR1 treatment (n = 8), and sham injury and vehicle treatment (n = 9).
Controlled cortical impact brain injury
The CCI TBI model is widely used in mice and produces a large cortical contusion and hippocampal damage ipsilateral to the injury, and also a degree of widespread neurodegeneration and axonal damage (Hall et al., 2008). All animals were anesthetized (sodium pentobarbital 65 mg/kg IP; Abbott Laboratories, North Chicago, IL for the NgR−/− study; and 1.4% isoflurane in a mixture of nitrous oxide and oxygen [70%/30%] delivered through a nose cone for the sNgR1 study), and placed in a stereotaxic frame adapted for mice (Kopf Instruments, Tujunga, CA), on a 37°C heating pad. An eye lubricant ointment was applied to protect the corneal membranes during surgery. After exposing the skull using a midline scalp incision, a 5-mm rounded craniectomy was performed over the left parietotemporal cortex between the lambda and the bregma, keeping the dura mater intact, according to the technique described previously (Murai et al., 1998). A CCI brain injury or sham injury was then performed as previously described (Israelsson et al., 2008; Murai et al., 1998). Briefly, CCI was induced using a piston set to compress the brain 1 mm at a speed of 5.0 m/sec (the NgR−/− study), or 0.5 mm at a speed of 2.8 m/sec (the sNgR study). The difference between the depth and speed of compression was due to local traditions at the two institutions, yet the CCI-induced tissue damage was markedly similar in both studies (see below). Sham-injured animals received the same anesthesia and all surgical procedures (craniectomy), but were not subjected to CCI brain injury. All animals were then allowed to recover from surgery on a heating pad maintained at 37°C. After the impact, the bone removed by the craniectomy was replaced and the scalp was sutured. The investigator performing the surgeries was blinded to the treatment status or genotype of each animal.
Neurological motor function
Neuroscore, NgR−/− study
Motor function was assessed in the NgR−/− study using the composite neuroscore (Murai et al., 1998), at baseline and at 48 h, and at 1, and 2, and 3 weeks post-injury by an observer who was blinded to animal status regarding injury or genotype. The animals were scored from 4 (normal) to 0 (severely impaired) by evaluating (1) forelimb function during walking on the grid and flexion response during suspension by the tail; (2) hindlimb function during walking on the grid and flexion/toe extension during suspension by the tail; and (3) resistance to lateral right and left pulsion. The maximum score for each animal was 12 (Longhi et al., 2005).
Rotarod and cylinder tests, sNgR1 study
In the sNgR1 study, we used the cylinder and rotarod tests to evaluate motor function. An accelerated rotarod (Panlab, Barcelona, Spain) turning at 4–40 rpm for a maximum of 5 min was used. Following pre-training twice daily for 2 days, the animals were tested on post-injury days 2, 7, 14, 23, and 33 (Hamm, 2001). On each test day, the animals were evaluated twice with a 5-min rest between trials, and the mean value of the two daily trials was used for analysis. Data are presented as percentages of the pre-injury values.
We also evaluated asymmetries in forelimb function using the cylinder test pre-injury (baseline), and at post-injury days 2, 7, 14, 23, and 33, according to established protocols (Schallert et al., 2000). The mice were placed in a 7.5-cm-diameter, 38-cm-high transparent cylinder and were filmed for 5 min or until 10 rears were observed. The films were later analyzed frame by frame at 10 frames/sec. Trials with less than five rears were excluded from further analysis.
Each frame in which at least one paw made contact with the cylinder wall was scored as either “contra,” “ipsi,” or “both.” This type of scoring relies on the amount of time each paw is used, rather than giving each rear a single score (Baskin et al., 2003). Performance was calculated as (both/2 + contra)/(contra + ipsi + both), as adapted from Woodlee and associates (Woodlee et al., 2005), and modified by replacing “ipsi” with “contra” in the numerator so that impaired performance gives a lower value. Data are presented normalized to the baseline, pre-injury performance.
Morris water maze, NgR−/− study and sNgR1 study
Evaluation of learning ability in mice following sham or CCI brain injury was performed at 4 weeks post-injury using the Morris water maze (MWM) test of learning and memory ability (Morris, 1984). The MWM has previously been shown to be a highly sensitive paradigm for measuring post-traumatic visuospatial learning and memory in mice (Longhi et al., 2005; Smith et al., 1995; Uryu et al., 2002). The MWM set-up used either a white circular pool 1 m in diameter (NgR1−/− study), or a pool 1.4 m in diameter (sNgR1 study), filled with water at 20°C. In the MWM, mice learn to escape from the water onto a platform submerged 0.5 cm using simple external visual cues. Each swim trial was run by placing the mouse in the tank at one of four designated entry points (W, N, E, and S) facing the wall, activating the video-based computer tracking system (Accutrak®, San Diego, CA for the NgR1−/− study, or HVS Image Ltd., Buckingham, U.K. for the sNgR1 study), and the trial was terminated when the mouse located the platform. Latencies to reach the platform were recorded for each trial, with a maximum of 60 sec per trial. The animal was allowed to remain undisturbed on the platform for 15 sec to acquire the visual cues surrounding the pool. In the NgR1−/− study, the learning task consisted of two daily trial blocks with four trials per block, for three consecutive days for a total of 24 trials. At 48 h following the last learning trial, the animals were evaluated in the maze for their ability to recall the previously learned task, the 60-sec probe (memory) trial, for which a memory score was calculated for each animal by multiplying the amount of time spent in the zones, weighted according to its proximity to the platform, according to a paradigm previously described in detail (Murai et al., 1998; Smith et al., 1991). The naïve NgR1−/− mice were evaluated in a separate experiment using the same pool diameter, water temperature (20°C), and trial block design as in the NgR−/− study.
In the sNgR1 study, the mice were subjected to four daily learning trials over four consecutive days followed by a 60-sec probe trial at 72 h following the last learning trial. The probe trial analyzed the latency to reach the platform area, and how many times the animals passed the area where the platform had been located in the previous trials. Following all MWM trials described in this section, including the memory probe trial, the animals were placed under a heating lamp to maintain normothermia.
Hemispheric tissue loss, NgR−/− study and sNgR1 study
Following behavioral evaluation, the animals were over-anesthetized with sodium pentobarbital (200 mg/kg, IP), and transcardially perfused with heparinized saline, followed by 4% paraformaldehyde (PFA) in PBS, and the brains were removed and post-fixed in PFA at 4°C. The brains from the NgR1−/− study were embedded in paraffin and the brain blocks were cut from bregma 1 mm to −4.5 mm in 5-μm-thick coronal sections on a microtome (Anglia Scientific, Cambridge, U.K.)
In the sNgR1 study, the animals were perfused with 50 mM sodium sulfide followed by 4% PFA in PBS to enable Timm stain evaluation (see below). The brains were post-fixed in 4% PFA, cryoprotected in 30 % (w/v) sucrose, flash-frozen in dry ice–chilled isopentane at −55°C, and cut into 25-μm sections using a cryostat (HM500; Microm GmbH, Walldorf, Germany). Sections were stained with Mayer's Hematoxylin (Histolab, Gothenburg, Sweden) and eosin (Histolab), and imaged using a digital scanner (Nikon Super CoolScan 4000ED; Nikon Imaging, Tokyo, Japan; NgR−/−study), or digitally photographed using a stereomicroscope (Zeiss Axio Imager Zl.; Zeiss Gmbh, Göttingen, Germany) with a digital camera (Axio Cam Mcm5c, Zeiss). The periphery of the contralateral and ipsilateral hemispheres were traced on each image by an evaluator blinded to the injury and treatment status of each animal, and the area of each hemisphere and cortical lesion volume were calculated using a calibrated image analysis routine. Six sections evenly distributed over the injury site between bregma 0 mm and −4.0 mm (Paxinos and Franklin, 2001) were used for the evaluation of hemispheric tissue loss. The hemispheric lesion volume between two bregma levels were calculated as d*(A1 + A2)/2, where d is the distance between sections, and A is the measured area (Zhang et al., 1998). Based on previous investigations that showed negligible contralateral tissue loss following experimental TBI, the contralateral hemisphere was used in each section to control for inter-animal variation in brain size (Zhang et al., 1998). Hemispheric tissue loss was calculated as a percentage of the contralateral (uninjured) hemisphere volume (Vc) using the following formula:
where Vi represents the volume of the ipsilateral (injured) hemisphere.
Timm stain, NgR−/− study
To evaluate sprouting of the zinc-containing mossy fibers in the hippocampus, Timm-stained sections were evaluated according to previously published protocols (Cavazos et al., 1991; Nissinen et al., 2001; Sloviter, 1982). Glasses were stirred in the dark in a developing solution containing 180 mL 50% (w/v) gum arabic, 30 mL 2 M citrate buffer, 90 mL 0.5 M hydroquinone, and 1.5 mL 1 M AgNO3, until sufficiently stained (approximately 1 h). The Timm reaction was inhibited by a gentle rinse (in the dark) for 30 min, and the glasses were then placed in 5 % (w/v) Na2S2O3 for 12 min, washed in H2O twice for 5 min each, dehydrated, and cover-slipped using Pertex (Histolab).
We then evaluated Timm staining in the ipsilateral and contralateral hippocampus at sections from −1.5 (two areas), −2.5 (three areas), and −3.5 mm (three areas) bregma, according to the protocol originally published by Cavazos and colleagues (Cavazos et al., 1991), in which the sections were scored from 0–5. Using this scale, “0” means no granules; “1” means sparse granules in the supragranular region and in the inner molecular layer; “2” means granules evenly distributed throughout the supragranular region and the inner molecular layer; “3” means an almost continuous band of granules in the supragranular region and inner molecular layer; “4” means a continuous band of granules in the supragranular region and in the inner molecular layer; and ”5” means a confluent and dense laminar band of granules that covers most of the inner molecular layer, in addition to the supragranular region.
β-amyloid immunohistochemistry, NgR−/− study and sNgR1 study
Several studies have demonstrated physical interactions between reticulon family proteins (including Nogo) and β-site APP-cleaving enzyme 1 (BACE-1), one of the proteases responsible for Aβ production from APP (He et al., 2004). Previously, NgR1−/− mice, bred onto a mouse transgenic model of Alzheimer's disease, influenced Aβ formation and plaque deposition (Park et al., 2006a, 2006b). Here, the sections were treated at +85°C for antigen retrieval (citrate buffer, 25 mM at pH 7.3) and rinsed in 1× PBS. The sections were immersed in 70% formic acid for 10 min followed by extensive rinsing in water. The sections were incubated in H2O2 (0.3%) in 50% Dako block/50% 1× PBS for 15 min, and permeabilized using 0.4% Triton X-100 for 5 min prior to application of primary antibody. Dako block was used to diminish unspecific binding and incubation with primary antibody, and a polyclonal Aβ40-specific antibody (Biosource, Camarillo, CA) was allowed to pursue overnight at 4°C. The sections were then incubated with secondary goat anti-rabbit antibody (Vector Laboratories, Burlingame, CA) for 30 min at room temperature, with streptavidin-HRP (Mabtech, Cincinnati, OH) for 30 min, and the reactions were finally developed with NOVA Red. The sections were washed, dehydrated, immersed in xylene, and mounted in dibutyl phthalate xylene for light microscopy. Hippocampal Aβ immunostaining was compared to separate sections from positive controls, APP-ArcSwe transgenic mice (Lord et al., 2006), known to form early intracellular and extracellular Aβ deposits, and to negative controls, APP-KO, expressing neither human nor mouse APP.
Statistical analysis
Continuous variables are presented with means ± standard error of the mean (SEM). Neuroscore is presented as median + 75th percentile, and Timm stain data are presented with box-plots showing the median, interquartile range, and min-max values. All data were evaluated for normal distribution and did not meet the assumptions for parametric analysis. Thus, non-parametric analyses were performed using a Kruskal-Wallis analysis of variance (ANOVA) on each evaluated time-point, and if significant, followed by Mann-Whitney U test. A p value of < 0.05 was considered significant. All data were analyzed using Statistica® (StatSoft, Tulsa, OK) software.
Results
Animals
There was no mortality in the NgR−/− study (n = 26). In the sNgR study, a total of 52 animals were anesthetized and subjected to CCI or sham injury. Of these mice, two died during surgery and four died of unknown causes, five were euthanized due to wound infections following surgery, and one brain-injured animal with a minimal cortical injury and no neurological motor deficit was excluded from the analysis. The mortality was not attributed to any treatment group. In total, 40 animals were included in the sNgR study.
Neurological motor function
Up to 4 weeks post-injury, we evaluated the effects of genetic deletion of or pharmacological neutralization of NgR1 on neurological motor function following CCI brain injury.
Neuroscore
TBI induced a significant deficit in neurological motor function in the NgR1−/− mice and WT littermates at 48 h and 1 week post-injury (p < 0.05; Fig. 1a). The median motor function scores of all groups of brain-injured mice gradually recovered over the 3-week observation period. There were no significant differences between the brain-injured NgR1−/− group and the brain-injured WT littermate group.

(
Rotarod
In the sNgR1 study, brain-injured mice, regardless of treatment status, performed worse on the rotarod tests than the sham-injured, vehicle-treated control mice only at post-injury day 2 (p < 0.05; Fig. 1b). In all groups, there was a marked improvement in rotarod latencies over the study period, which ultimately equaled or exceeded the pre-injury performance. Treatment with sNgR1 did not significantly alter rotarod performance, although there was a trend toward improved recovery in the sNgR1-treated brain-injured group.
Cylinder test
Asymmetry in forelimb use during spontaneous rearing was evaluated using the cylinder test in the sNgR1 study. Brain-injured, vehicle-treated animals used their contralateral paw significantly less than the sham-injured, vehicle-treated mice for each evaluated post-injury time point (p < 0.05; Fig. 1c). The average use of the contralateral paw remained similar at all evaluated post-injury time-points, and no recovery of function was observed in either brain-injured group. Treatment with sNgR1 did not significantly alter forelimb use in the cylinder test.
Morris water maze
Four weeks following CCI brain injury in the NgR−/− and sNgR studies, the animals were evaluated for their ability to learn the position of a hidden platform in the MWM. In both studies all animals were able to swim without alterations in swimming ability or swim speed (data not shown).
NgR1−/− study
The brain-injured, WT littermate mice had significantly longer latencies to locate the hidden platform at trial blocks 1, 2, 4, and 6 (p < 0.05; Fig. 2a), and the brain-injured NgR1−/− mice were significantly worse at trial blocks 1–6 (p < 0.05) than sham-injured controls. Moreover, the brain-injured NgR1−/− mice showed consistently longer latencies to locate the platform than the brain-injured WT control mice in trial blocks 3–6 (p < 0.05 at trial block 3). In contrast, naïve NgR1−/− mice quickly learned the MWM task with no significant differences from the littermate controls (Fig. 2b), and there was no difference in probe trial performance (data not shown). At 48 h following the last MWM trial, the platform was removed and the probe (memory) trial was performed. Brain-injured, NgR1−/− mice had lower memory scores compared to sham-injured controls (p < 0.05; Fig. 2c), and showed a non-significant trend toward a lower memory score compared to the brain-injured, WT littermate controls.

(
sNgR1 study
The sham-injured animals quickly learned the MWM task, whereas the brain-injured, vehicle-treated animals had slightly longer latencies to find the hidden platform than vehicle-treated, sham-injured controls (Fig. 2d). However, sNgR1-treated, brain-injured mice had longer latencies than brain-injured, vehicle-treated animals at days 28–30 post-injury (p < 0.05 at day 28, p = 0.10 at day 29, and p = 0.08 at post-injury day 30). The number of timed-out trials (failure to locate the platform) was also higher in the sNgR1-treated, brain-injured mice than in all other injury groups (p < 0.05; data not shown). At 72 h following the last MWM trial, the platform was removed for the probe (memory) trial. Brain-injured animals had a longer latency to reach the platform area and a reduced number of platform passes than sham-injured controls (Fig. 2e). Brain-injured, sNgR1-treated mice showed a trend toward longer latencies to reach the platform area than the brain-injured, vehicle-treated controls (Fig. 2e), and a reduced number of passes over the platform area (p = 0.06; data not shown).
Tissue loss
We evaluated TBI-induced tissue loss in the injured ipsilateral hemisphere, and the loss of tissue was markedly similar for the genetic and pharmacological experiments (Fig. 3a–c). In both studies, CCI induced a significant loss of tissue in the injured hemisphere at 4 weeks post-injury compared to sham-injured controls (p < 0.05), but there were no significant difference between the brain-injured groups, regardless of genotype or pharmacological treatment status (Fig. 3a).

(
Timm staining
Mossy fiber sprouting is observed during epileptogenesis and has been observed following TBI in the rat (Kharatishvili et al., 2006) and mouse (Hunt et al., 2009), and was selected as a measure of hippocampal sprouting in the sNgR1 study. We evaluated eight areas ipsilateral and contralateral from bregma −1.5 to −3.5 mm (Fig. 4a). The sum of the Timm scores from the eight regions in each hemisphere is presented as the total Timm score in Figure 4c. When compared to sham-injured, vehicle-treated mice, brain-injured mice showed increased ipsilateral Timm staining in the majority of the evaluated areas (p < 0.05; Fig. 4b–c). Specifically, in both treatment groups, TBI caused an extra band of black granules in the supragranular areas (Fig. 4b) that was more marked on the ipsilateral side. We hypothesized that sNgR1 treatment might alter injury-induced changes in the pattern of Timm staining. There was a significantly increased amount of granules in three areas on the ipsilateral side in sham-injured, sNgR1-treated animals compared to vehicle-treated, sham-injured control mice (p < 0.05; Fig. 4d). Although treatment with sNgR1 increased Timm staining in area h (p = 0.07; Fig. 4e), there was not a significant increase in Timm stain with sNgR1 treatment in most evaluated areas of brain-injured animals. Thus, brain injury robustly induced hippocampal sprouting as detected by Timm staining, and sNgR1 treatment slightly increased sprouting in sham-injured, but not in brain-injured, animals.
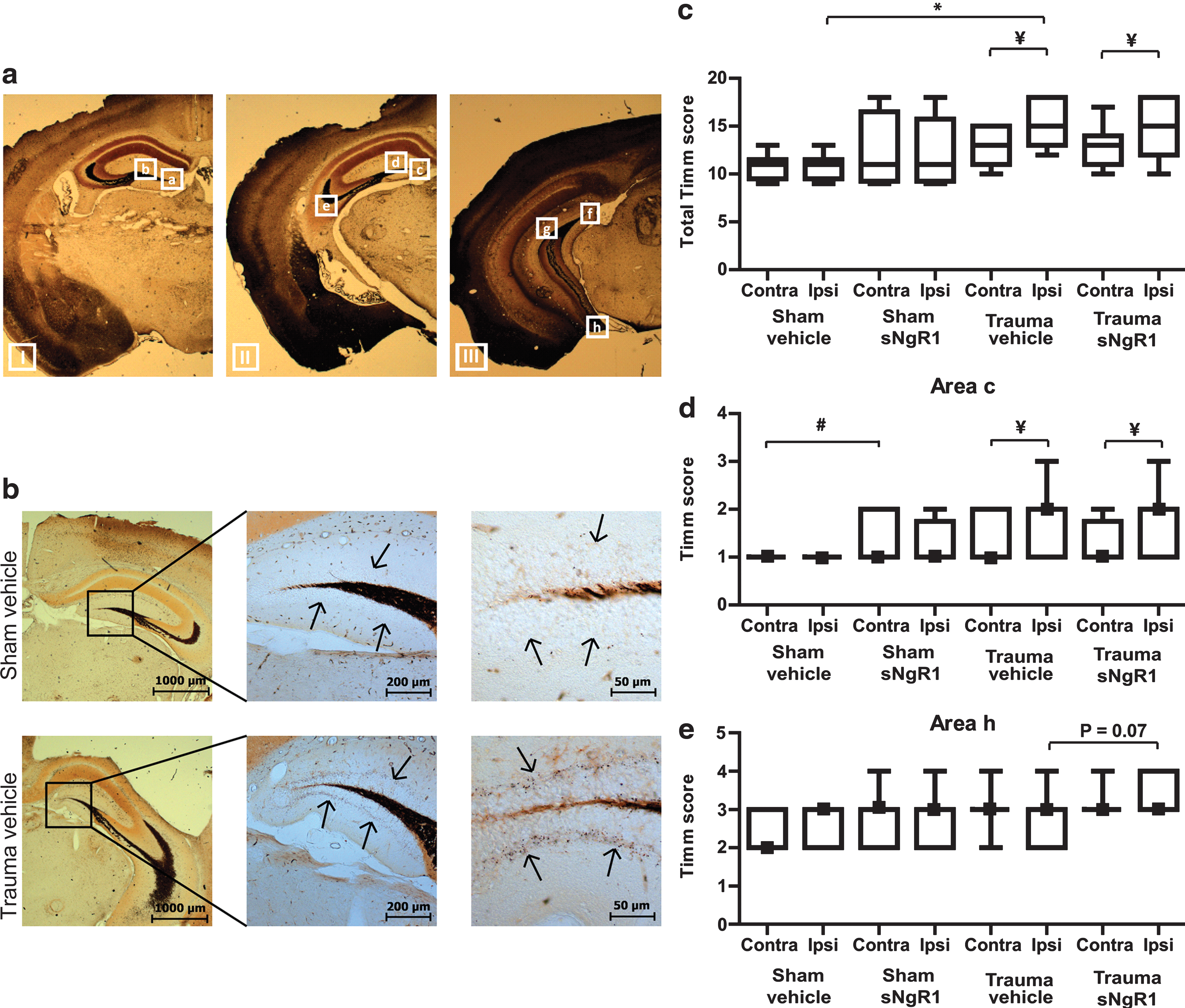
(
Aβ immunohistochemistry
We attempted to elucidate the potential mechanism responsible for the unexpected cognitive results and evaluated β-amyloid (Aβ) immunohistochemistry in the hippocampus of the brain-injured animals. In positive control mice, APP-ArcSwe transgenic mice (Lord et al., 2006), an Aβ40-specific antibody stained intracellular Aβ and extracellular Aβ plaques in brain tissue (Fig. 5a and b). We evaluated the pyramidal cell layer of the hippocampal CA1 field, and observed only a few scattered intracellular Aβ-immunoreactive aggregates, similar to those seen brain-injured NgR1−/−, WT controls, sNgR1-treated, and vehicle-treated mice (Fig. 5c–d). Thus there was no or very little accumulation of Aβ as a result of TBI in these mice, and Aβ-immunostaining was not significantly altered by sNgR1 treatment. No Aβ plaques were detected in the negative control section from APP-KO mice (Fig. 5e–f).
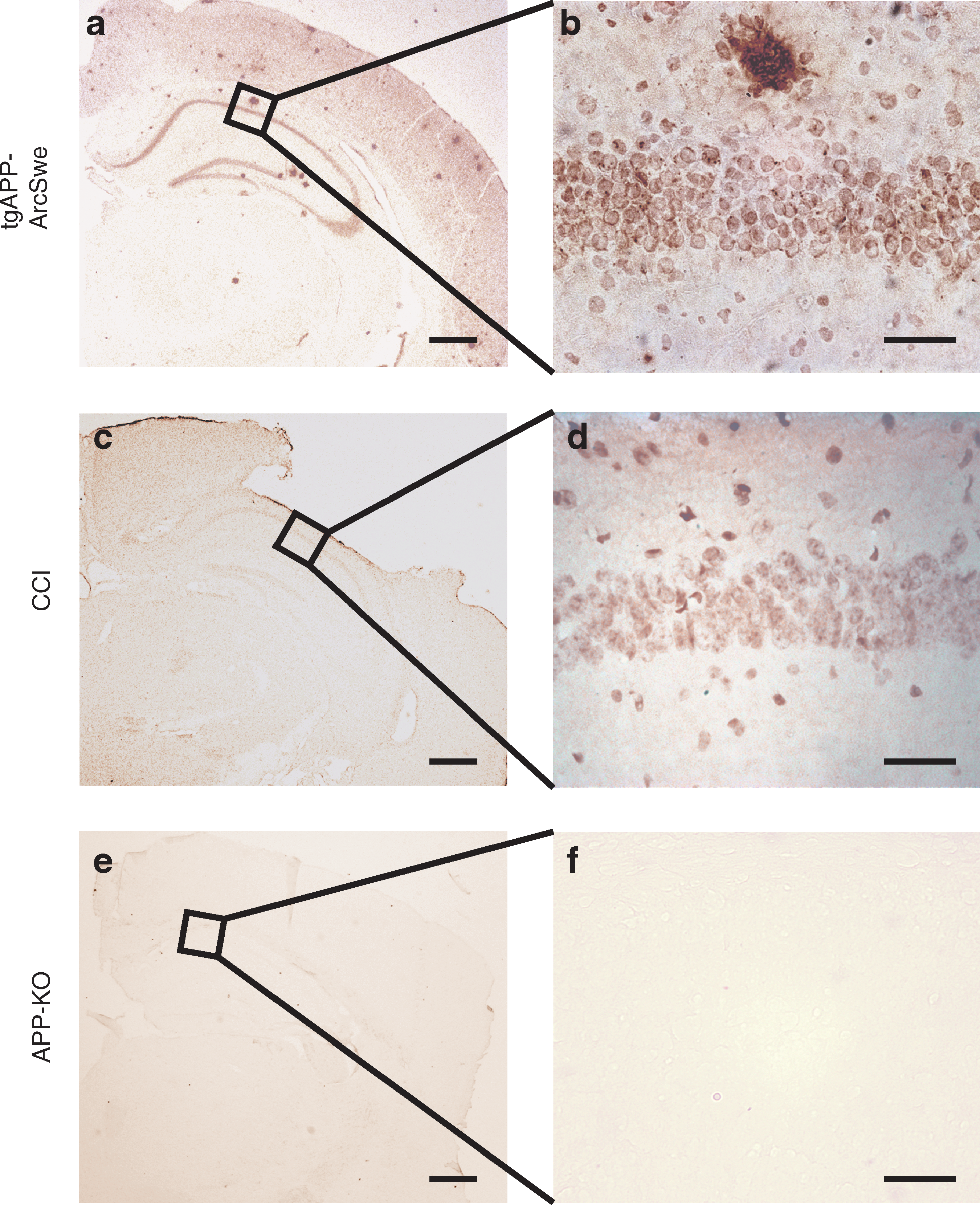
β-Amyloid (Aβ) immunohistochemistry, sNgR1 study, at 20 × (
Discussion
The major finding of the present report is that both Nogo-66 receptor-1 (NgR1)-deficient mice and wild-type mice receiving pharmacological neutralization of NgR1 were impaired in a visuospatial cognitive task following focal traumatic brain injury (TBI), compared to their wild-type, littermate, or vehicle-treated controls. Additionally, neither genetic absence of NgR1 nor pharmacological neutralization of NgR1 influenced the neurological motor deficits associated with this TBI model. Our data also demonstrate that hippocampal sprouting was increased by TBI and by sNgR1 treatment in sham-injured animals. The present report is the first to evaluate genetic absence and pharmacological neutralization of NgR1 following TBI, and provides novel information about the role of myelin inhibitors in cognitive function following TBI.
Neither genetic deletion nor pharmacological neutralization resulted in any change of post-injury neurological motor function. A degree of hypoactivity and motor impairment have been reported in uninjured, naive NgR1−/− mice (Kim et al., 2004). We cannot exclude the possibility that the NgR1−/− mice used in the present report had subtle pre-injury motor deficits that were undetectable using our motor function tests. However, no pre-injury deficits in neurological motor function as assessed using the neuroscore was detected, suggesting that any pre-injury deficit was mild and did not substantially contribute to the present results. Previously, improved functional motor recovery was correlated with increased rubrospinal plasticity after ischemic brain injury in NgR1−/− mice (Lee et al., 2004; Zheng et al., 2005), although NgR1 may not be important for the regeneration of all efferent pathways following CNS injury (Kim et al., 2004). It is important to note that the motor recovery was nearly complete in the studies detailed here, and the major deficits were in memory function. Since our tests of motor function mainly assess corticospinal tract function, other testing of neurological motor function may have yielded different results.
In contrast to the present study, pharmacological inhibition of NgR1 in other models of CNS injury has consistently resulted in an improvement of neurological motor function when administered in a subacute or delayed fashion (see Introduction). In these reports, the enhanced recovery was associated with increased axonal regrowth and/or sprouting of uninjured axon fiber tracts post-injury. Here, we used PBS as a vehicle control treatment, as previously reported by others (Harvey et al., 2009; Wang et al., 2006). Although unspecific reactions may occur from the administered IgG-Fc part of the fusion protein, the results were similar when using PBS vehicle to those of other reports using control IgG treatment in other models of CNS injury (Lee et al., 2004; Li et al., 2004; Park et al., 2006b). Since the behavioral results were markedly similar in both studies, our data suggest that reduced NgR1 signaling is responsible for the observed behavioral results, and argue against an unspecific effect of the drug treatment. The CCI brain injury model used in the present report is primarily a focal TBI model with marked tissue destruction at the site of impact, predominantly in the ipsilateral cortex and hippocampus (Dixon et al., 1991; Smith et al., 1995). However, this TBI model also shows widespread axonal damage in brain areas remote from the impact (Hall et al., 2008). The rationale for pharmacologically targeting NgR1 was twofold: first, NgR1 inhibition may induce an enhanced axonal regeneration in damaged white matter tracts, and secondly, it may also increase plasticity in relatively uninjured areas, which has been shown to occur in other disease models (Lee et al., 2004; McGee et al., 2005). The explanation for our unexpected results remain unclear, although the widespread diffuse axonal injury components present in the CCI injury model (Hall et al., 2008) may impair the potential for recovery of the contralateral hemisphere. In view of our findings, targeting NgR1 in a diffuse axonal injury model may help clarify the role for NgR1 in TBI.
We have shown previously that no significant changes in cortical or hippocampal NgR1 expression occur following experimental TBI in the rat, in contrast to Rho activation (Dubreuil et al., 2006; Marklund et al., 2006b). The role for NgR1 signaling in TBI has not been established, and in the present report, we observed an exacerbation of learning and memory deficits following TBI in NgR1−/− mice compared to similarly brain-injured WT littermates and following sNgR1 administration. NgR1−/− mice have a compensatory upregulation of Nogo-A protein in oligodendrocytes (Kim et al., 2004), and the impaired cognitive function may not be due solely to the deletion of the NgR1 gene, since Nogo-A may interfere with hippocampal function and cognitive recovery (Meier et al., 2003; Mingorance et al., 2004). Conversely, Nogo-A (beginning 24 h post-injury) inhibitory antibodies significantly improved cognitive performance following fluid percussion TBI in the rat (Lenzlinger et al., 2005; Marklund et al., 2007), without influencing hippocampal or cortical cell loss (Lenzlinger et al., 2005; Marklund et al., 2007). Similarly to reports following cerebral ischemia in rats or mice (Lee et al., 2004; Wang et al., 2006), we observed no difference in hemispheric tissue loss among the brain-injured groups in both studies detailed here. Together, these results suggest that NgR1 is not involved in the post-injury cascades contributing to TBI-induced cortical or hippocampal cell death. In the present report, naïve NgR−/− mice learned the MWM task similarly to littermate control mice, suggesting that TBI-induced factors are responsible for the observed cognitive deficits in NgR1−/− mice. Next, we sought to determine the molecular mechanisms behind the cognitive impairment.
First, we evaluated accumulation of the amyloid precursor protein (APP) metabolite Aβ in the neuropil. There are known interactions between reticulon family proteins and BACE1, and NgR1−/− mice bred to a mouse transgenic Alzheimer's disease model showed enhanced Aβ deposition (Park et al., 2006a). Thus, we evaluated Aβ by Immunohistochemistry, and found only non-distinct intracellular inclusions of Aβ in brain-injured mice, regardless of mouse genotype. Following TBI in the mouse, no or only minimal increases of Aβ have previously been observed (Smith et al., 1998), and our results suggest that accumulation of Aβ deposition is not a major contributor to this TBI model, nor is Aβ the likely cause for the impaired cognitive function observed in our report.
Secondly, hippocampal (Scheff et al., 2005) and corticospinal (Lenzlinger et al., 2005; Marklund et al., 2007) plasticity has been demonstrated following experimental TBI. We used the Timm stain to evaluate mossy fiber sprouting, which is observed in patients with temporal lobe epilepsy and in animal models of epilepsy. Timm staining is increased and correlates with seizure activity over the long term following TBI in the rat (Kharatishvili et al., 2006), and in the mouse (Hunt et al., 2009). Additionally, seizures induced by kainic acid administration result in a decreased expression of NgR1 mRNA, supporting a link between NgR1 expression and a response to seizures (Josephson et al., 2003). Here we used Timm stain in eight different hippocampal regions as a measure of TBI-induced hippocampal sprouting. Although minimal mossy fiber sprouting was already evident at 7 days post-TBI (Hunt et al., 2009), the exact location of and the time course for mossy fiber sprouting following TBI in the mouse remain to be defined. We observed that TBI alone markedly increased Timm staining bilaterally. sNgR1 treatment caused increased bilateral hippocampal sprouting in sham-injured animals, suggesting that NgR1 inhibition influences mossy fiber sprouting, although these effects were not detected in brain-injured animals, except for a non-significant trend in one of the evaluated hippocampal regions. Thus our results indicate that NgR1 inhibition may influence mossy fiber sprouting, but also indicate that other mechanisms contributed to our results. The sprouting response to TBI is multifactorial, and TBI itself may induce numerous molecular changes related to plasticity, including increased GAP-43 expression (Marklund et al., 2007).
Supranormal plasticity may occur in the absence of NgR1 (McGee et al., 2005), and we hypothesize that the absence of NgR1 is associated with aberrant sprouting or enhanced plasticity that may interfere with the cognitive recovery process post-injury. Conversely, NgR1 inhibition was associated with an increased expression of the axonal growth-promoting protein SPRR1A (Li and Strittmatter, 2003). These results indicate that other factors, induced by TBI, may be involved in the sprouting response and recovery process that may be influenced by NgR signaling. Additionally, downregulation of NgR1 occurs during learning, and is suggested to be required for the formation of long-term memory (Josephson et al., 2003). Inducible overexpression of NgR1 in forebrain neurons was also recently shown to be associated with impaired long-term memory, implying a complex role for NgR1 in memory consolidation (Karlen et al., 2009). Neither NgR1 deletion nor sNgR1 treatment impaired memory function in naive or sham-injured mice in our studies, although we cannot exclude the possibility that other, more complex tests could have revealed subtle deficits in cognitive function induced by the absence of NgR1. However, our data accentuate the unique and complex nature of change in brain function after TBI, and emphasize the need for targeted therapies.
In conclusion, our data suggest that both inhibition of NgR1 signaling and genetic deletion of NgR1 were associated with markedly similar cognitive deficits at 4 weeks post-injury, in contrast to the cognitive improvement observed with Nogo-A inhibition following TBI in the rat. For practical reasons, there were some aspects of the experimental design that differed among the departments performing the experiments. We prefer to emphasize the similarity in the findings from both the genetic deletion and pharmacological neutralization studies. Further studies evaluating compensatory molecular changes may clarify the mechanisms responsible for the cognitive impairment following TBI in NgR1−/− mice. Neutralization of NgR1 using other dosing regimens and/or time windows, in addition to evaluation using other TBI models and species, are needed to further define the role for NgR1 inhibition following experimental TBI.
Footnotes
Acknowledgments
We thank Diego Morales, Scott Fujimoto, Rishi Puri, Carl T. Fulp, Görel Lindman, David LeBold, Lena Rundström, Caroline Hagbohm, and Hilaire J. Thompson for excellent technical support. The Uppsala University Transgenic Facility (UUTF ) is greatly acknowledged for their help in developing the APP transgenic mouse models used in this study.
Supported by Upplandsstiftelsen and the Swedish Brain Foundation (to N.M.), Uppsala University (to N.M. and L.H.), Swedish Research Council (to N.M. and L.H.), Jeanssons Foundation (to N.M.), National Institutes of Health (NIH) NS RO1-40978 (to T.K.M.), NIH NS RO1-56485 (to S.M.S.), NIH NS RO1-42304 (to S.M.S.), a Merit Review Grant from the Veterans Administration (to T.K.M.), and NIH NS P50-08803 (to T.K.M.).
Author Disclosure Statement
No competing financial interest exist.