
Abstract
Glial scar formation occurs in response to brain injury in mammalian models and inhibits axonal growth. Identification of molecules that may mediate reactivity of astrocytes has become a leading therapeutic goal in the field of neurotrauma. In adult rodent brain and spinal cord, many of the Eph receptors and their ephrin ligands have been demonstrated to be upregulated on reactive astrocytes at the injury site; however, little is known about the expression of these molecules in nonhuman primate injury models. This study examines the role of the tyrosine kinase EphA4 receptor, which predominantly binds most ephrin ligands, after injury in marmoset monkey brain. Following lesioning of the primary visual cortex (V1) in the adult marmoset, EphA4 is strongly upregulated on reactive astrocytes around the lesion site, which secrete extracellular matrix molecules such as chondroitin sulfate proteoglycans, which are known for their inhibitory effect on axonal growth and regeneration. This astrocyte reactivity was also associated with neuronal death in the area adjacent to the lesion site. EphA4 activation induced by clustered ephrin A5-Fc–mediated astrocyte proliferation and glial fibrillary acidic protein expression in vitro, as demonstrated by closure of scratched wound and MTT assays, occurs via two potential signaling pathways, the mitogen-activated protein kinase and Rho pathways. These results in a nonhuman primate model highlight the importance of developing pharmacotherapeutic approaches to block these molecules following brain injury.
Introduction
After a CNS injury, astrocytes become reactive and hypertrophic and upregulate expression of intermediate filaments, such as nestin and vimentin, and glial fibrillary acidic protein (GFAP) (Buffo et al., 2008; Pekny and Nilsson, 2005). In their reactive state astrocytes proliferate and migrate to the injury site to form the glial scar, and release factors that mediate the tissue inflammatory response and remodeling after injury (Ridet et al., 1997; Silver and Miller, 2004). The glial scar consists predominantly of reactive astrocytes, which secrete chondroitin sulfate proteoglycans (CSPGs) into the extracellular matrix. These molecules are known for their inhibitory effects on neuronal re-growth (Galtrey and Fawcett, 2007; Lin et al., 2008; McKeon et al., 1991; Snow et al., 1990). A key role for EphA4 in mediating scar formation and axonal regeneration was demonstrated using a spinal cord injury model in EphA4-deficient mice (Goldshmit et al., 2004). Following spinal cord hemisection, scar formation was significantly reduced in EphA4-null mice, while in wild-type mice EphA4 was upregulated on the astrocytes. Not only was GFAP expression reduced in the astrocytes of EphA4-null mice, but CSPG staining was also reduced, suggesting that other major axonal growth inhibitors may be decreased in the scar. Furthermore, astrocytes from EphA4-deficient mice exhibit attenuated responses to inflammatory cytokines in vitro, suggesting a central role for EphA4 in astrocyte reactivity.
Most studies to date examining Eph receptor expression during development and post-injury have been performed in rodent models; however, there are several differences between rodent astrocytes and those found in the primates (including humans). For example, some inflammatory enzymes, such as nitric oxide synthase, are produced by microglia in rodents and by astrocytes in humans (Zhao et al., 1998). In addition to the fibrous and protoplasmic astrocytes found in rodents, humans also have interlaminar and polarized astrocytes (Oberheim et al., 2006). Moreover, primate protoplasmic astrocytes are more complex and larger than those observed in rodents (Oberheim et al., 2006). In light of these differences, it is important to determine whether primate astrocytes respond to injury in a similar manner to rodent astrocytes. Expression of Ephs and ephrins in the nonhuman primate brain has been demonstrated at the mRNA level during cortical development (Donoghue and Rakic, 1999; Sestan et al., 2001), and in the adult brain (Hafner et al., 2004; Xiao et al., 2006). Furthermore, one study demonstrated upregulation of the Ephs and ephrins in humans with multiple sclerosis, predominantly on the reactive astrocytes in demyelinated areas (Sobel, 2005).
It is becoming increasingly evident that the modalities and dynamics of the astrocytic response to damage are crucial to the outcome of brain injury and the degree of neurological damage. Therefore, manipulation of astrocyte reactivity may represent an important avenue for therapeutic intervention after brain injury. In this present study we examined EphA4 expression after cortical injury (lesion) of the primary visual cortex (V1) of the adult nonhuman primate; a lesion that is commonplace following penetrative head trauma. A combination of in vivo and in vitro techniques were used to determine whether this receptor is associated with astrocytic gliosis in primate brain in a similar way to rodent models.
Methods
Animals
Three young adult (12-month-old) marmoset monkeys (Callithrix jacchus) weighing ∼350 g were used in this study. The animals were housed in family groups (12/12-h light/dark cycle, temperature 31°C, humidity 65%). All experiments were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and were approved by the Monash University Animal Ethics Committee, which also monitored the welfare of these animals.
Visual cortex ablation
The animals were initially anesthetized with intramuscular injections of Alfaxan® (12 mg/kg−1) and diazepam (3.0 mg/kg−1). After 15 min they were given antibiotic (procaine penicillin 25 mg/kg−1 IM) and the anesthesia was maintained with isoflurane (1–2% in 0.5 L/min−1 oxygen). Each animal was placed in a stereotaxic frame on a thermostatically controlled heating pad to maintain core body temperature at 38°C, and blood oxygen level, heart rate, and temperature were monitored continuously throughout the experiment. A small incision was made following the midline of the cranium, and an opening (6 × 8 mm) was made over the occipital pole (the back end of the cranium, where area V1 is located) using a burr drill bit, cooled with drops of iced saline. The exposed dura mater was kept moist with topical application of saline, and resected. Once the target area, V1, was visualized, a fine-tip surgical electrocautery device was used to lesion the opercular region of V1, approximately 5 mm3 in volume. Once the lesion was completed, a piece of saline-infused Gelfoam (Pharmacia & Upjohn Inc., Bridgewater, NJ) was inserted into the empty space of the lesion, the dura mater was replaced over the cortex, and a piece of Gelfilm (Pharmacia & Upjohn Inc.) used to cover the incised dura. The bone fragment was replaced and bonded with Vetbond® (3M, St. Paul, MN). The skin incision was then sutured closed, and a topical local anesthetic gel (xylocaine 5% cream) was applied to the incision site to minimize discomfort in the period immediately after surgery.
Tissue preparation
Three weeks post-injury, the marmosets were deeply anesthetized with an overdose of sodium pentobarbitone (100 mg/kg−1 IP) and transcardially perfused with 0.1 M heparinized phosphate buffer (PB; pH 7.2), followed by 4% paraformaldehyde in PB. Cerebral tissues were immediately removed and post-fixed for 24 h in 4% paraformaldehyde in PB containing 10% sucrose. Cryoprotection was achieved by serially transferring the tissue through solutions of 20% and 30% sucrose in PB, before slow freezing with liquid nitrogen (Warner et al., 2010). The hemispheres were left connected and five series of coronal or sagittal sections 40 μm thick were cut with a cryostat.
Immunohistochemistry
Sections were examined using standard immunohistochemical procedures to determine expression and localization of different proteins at the lesion site. After three 10-min washes in phosphate-buffered saline (PBS; pH 7.2), free-floating sections were incubated in PBS containing 0.3% Triton X-100 (PBS-TX) for 1 h, followed by blocking solution (PBS-TX containing 5% normal goat serum, NGS; Invitrogen, Carlsbad, CA) for 1 h at room temperature. Primary antibodies were diluted in blocking solution and incubated with sections overnight at 4°C. The sections were then washed three times in PBS and incubated with secondary antibodies for 2 h at room temperature, followed by three 20-min washes in PBS. The sections were then mounted using Fluoromount (Dako North America, Inc., Carpenteria, CA).
The primary antibodies used in this study included rabbit anti-EphA4 (1:300, prepared against a peptide corresponding to amino acids 938–953 of the intracellular Sterile Alpha Motif [SAM] domain of EphA4, obtained from Dr. J. Coonan, Walter and Eliza Hall Institute, Melbourne, Australia). This antibody showed no staining in EphA4–/– mouse tissue (Goldshmit et al., 2004). The EphA4 receptor structure is very conserved between species, such as mouse, human, and chick, including the SAM domain, which this antibody was made against. This region possesses 95% homology between mice and humans, as demonstrated using the ClustalW2 program from EMBL-EBI (Larkin et al., 2007; mouse- VGDWLQAIKMDRYKDNF TAAGYTTLEAVVHMSQDDLAR; human- VGDWLQAIK MDRYKDNFTAAGYTTLEAVVHVNQEDLAR). In addition, homology has been assessed by four methods of orthology prediction (INPARANOID, ORTHOMCL, ensembl compara, and Treefam) from the COMPARE database (Salgado et al., 2008); mouse anti-GFAP (1:1000; Invitrogen); mouse anti-NeuN (1:500; Millipore, Billerica, MA); rabbit anti-activated caspase-3 (1:500, BD Biosciences Pharmingen, Franklin Lakes, NJ); and mouse anti-CSPG (1:200; Sigma-Aldrich Co., St. Louis, MO).
For double immunofluorescence labeling, secondary antibodies included: goat anti-rabbit Alexa Fluor-488 (1:600; Molecular Probes, Eugene, OR), and goat anti-mouse Alexa Fluor-594 (1:800; Molecular Probes). When single antibodies were applied, the secondary antibody that was used was HRP-conjugated goat anti-rabbit IgG (Invitrogen), followed by 1–2 min of incubation with a metal-enhanced diaminobenzidine (DAB) substrate kit (Immunopure®; Pierce Protein Research Products, Rockford, IL).
Histology
Nissl-substance labeling was achieved by using a 0.1% cresyl violet solution. Slide-mounted sections were stained until layer 6 was discernible from the other layers (Bourne and Rosa, 2006).
Microscopy
Sections were examined by brightfield or fluorescence microscopy using a Axioplan Z1 (Zeiss, Berlin, Germany) epifluorescence microscope. Photomicrographs (1300 × 1030 dpi) were obtained with 2.5 × and 5 × Plan-Neofluar objectives (Zeiss), and acquired as digital images using an AxioCam (Zeiss) digital camera with AxioVision software (v. 4.4; Zeiss). Images were cropped and sized using Adobe Photoshop 11 and Illustrator 14. To confirm co-localization between caspase-3 and NeuN immunolabeling, optical sections of the sample were acquired on a Axoioplan Z1 (Zeiss) microscope equipped with the Apotome module and a 40 × objective, using AxioVision software.
Astrocyte culture
Primary astrocyte cultures were derived from the ablated area of post-natal day (PD) 14 marmoset monkey V1. Meninges were removed and the tissue was mechanically dissociated using a scalpel blade in minimal essential media (MEM; Gibco, Carlsbad, CA). The tissue was then incubated for 20 min in solution C (476 mg HEPES, 40 mg EDTA, 50 mg trypsin and, 2 mL of 1 mg/mL DNase I in 200 mL of calcium- and magnesium-free heparin buffered saline solution, adjusted to pH 7.6, filtered, sterilized, and stored at −20°C), followed by the addition of an equal volume of solution A (14 mg of trypsin inhibitor, 1 mL of 1 mg/mL DNase I dissolved in 100 mL MEM, filtered, sterilized, and stored at −20°C). After 1 min, the cell suspension was diluted using MEM (1:10), then pelleted, resuspended in Dulbecco's modified Eagle's medium (DMEM, Gibco) containing 10% FCS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and plated into 75-cm flasks. To enrich the mixed glial culture for astrocytes, the flasks were shaken on an orbital shaker at 150 rpm for 4 h. The growth medium was replaced after the shaking process, and the cells were cultured for 1 week before use. At this point, the cultures were trypsinized and re-plated on poly-D-lysine-coated chamber slides or into 10-cm Petri dishes at 1 × 104 cells/well or 1 × 106 cells/plate, respectively.
Analysis of signal transduction
To examine the different signaling pathways that are mediated through EphA4 receptor activation, the astrocytes were plated with or without specific factors as listed below. The cells were serum starved for 24 h, then factors were added for the indicated times. In experiments using Ephrin-A5-Fc for EphA4 receptor activation, 1.5 μg/mL of the peptide was pre-clustered for 30 min with 0.15 μg/mL anti-human IgG (Vector Laboratories, Burlingame, CA) at room temperature. Leukemia-inhibiting factor (LIF; Chemicon International, Temecula, CA) was added to a final concentration of 1000 U/mL. The cells were washed with cold PBS and lysed with lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA pH 8, 1.5 mM MgCl2, 2 mM PMSF, 0. 2mM Na3VO4), and a protein inhibitor cocktail (Roche Diagnostics Corp., Indianapolis, IN). The lysates were then clarified by centrifugation.
For analysis of EphA4 phosphorylation, the lysates were incubated for 2 h with anti-mouse-IgG agarose beads (Sigma-Aldrich) conjugated with mouse anti-phosphotyrosine (1:50; Cell Signaling Technology, Inc., Beverly, MA). Immunoprecipitates were collected by centrifugation, washed three times with HNTG buffer (20 mM HEPES pH 7.50, 150 mM NaCl, 0.1% TritonX-100, and 10% glycerol), resuspended in 35 mL 4 × loading buffer (200 mM Tris-HCl pH 6.8, 8% SDS, 0.04% bromophenol blue, 40% glycerol, and 400 mM DTT), and heated for 5 min at 100°C.
For Western blotting, protein samples were mixed with a 1:4 volume of 4 × loading buffer (200 mM Tris-HCl pH 6.8, 8% SDS, 0.04% bromophenol blue, 40% glycerol, and 400 mM DTT), and heated for 5 min at 100°C. Proteins were resolved by SDS-PAGE using 8–16% gradient polyacrylamide gels (Gradipore, Frenchs Forest, N.S.W., Australia) in running buffer (25 mM Tris, 200 mM glycine, and 0.1% SDS), and electrophoretically transferred onto a nitrocellulose membrane (0.45 μm; Bio-Rad Laboratories, Inc., Hercules, CA) in transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol). The membranes were blocked for 2 h in Tris-buffered saline (TBS; 0.05 M Tris pH 7.5, 0.15 M NaCl, and 0.1% Tween-20) containing 6% skim milk powder, and incubated with primary antibodies overnight, followed by a secondary antibody linked to HRP-anti-rabbit IgG (1:1000, Cell Signaling). Immunoreactive bands were detected with an enhanced chemiluminescence reagent (Pierce Protein Research Products). Total EphA4 expression levels were determined using rabbit anti-EphA4 antibody (1:2000; Upstate Biotechnology, Lake Placid, NY).
For mitogen-activated protein kinase (MAPK) activation, Western blots prepared using 20 μg of total protein lysate were probed using rabbit anti-p-MAPK (1:1000; Cell Signaling). The membranes were stripped using glycine solution (0.2 M pH 2) at 60°C for 10 min, followed by three washes in TBST and incubation with rabbit anti-MAPK (1:1000; Cell Signaling).
Rho GTPase activation was detected using the Rhotekin Rho Binding Domain (RBD) assay according to the manufacturer's instructions (Upstate Biotechnology). After treatment, the cells were washed once with ice cold PBS, then lysed in 0.5 mL Rho assay lysis buffer. The lysates were collected into a microcentrifuge tube, mixed briefly, and incubated on ice for 5 min. The lysates were then centrifuged at 14,000g at 4°C for 15 min. Endogenous Rho-GTP was precipitated from the cell lysates at 4°C for 1 h, using 30 μg of Rhotekin RBD-agarose for each sample. The beads were pelleted by centrifugation at 14,000g for 2 min, washed three times with the lysis buffer, and resuspended in 4× loading buffer. The eluted protein samples were resolved on 12% SDS gels and analyzed by Western blot. Rho was detected using rabbit anti-Rho (1:1000; Cell Signaling).
Statistical analysis
Quantitative analysis of detected bands was carried out by densitometric analysis of scanned blots using Image J software (NIH). Statistical comparison was made against normalized data, for which basal protein activity values in control untreated cells was set to 100%. Individual comparisons between specific treatments and untreated controls were made using a paired t-test. A p value of < 0.05 was considered significant.
Cell proliferation assay
The [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl]tetrazolium bromide (MTT) assay (Sigma-Aldrich), which determines mitochondrial activity in living cells, is commonly used as a proliferation assay (Mosmann, 1983). Living cells transform the tetrazolium ring into dark blue formazan crystals, which can be quantified by reading the optical density (OD). An increase in OD correlates with an increase in cell number over time. Marmoset monkey astrocytes were plated on 96-well plates at 3 × 103 cells/well in DMEM supplemented with 10% FCS in the presence or absence of LIF (1000 U/mL), clustered ephrinA5-Fc or ephrinA2-Fc (1.5 μg/mL). The MTT assay was performed at 2, 24, 48, and 72 h after plating. At each time point cells were incubated with MTT (0.25 mg/mL) for 2 h at 37°C. The cells were then lysed with an equal volume of acidic isopropanol (0.04 M HCL in absolute isopropanol) and the OD of the formazan product was measured at 550–650 nm (550 excitation/650 absorbance). Data are expressed as the mean ± SEM (6 replicate wells in each experiment; n = 3). When comparing two averages, the Student's t-test assuming equal variance was employed; p < 0.05 was considered statistically significant.
Astrocyte scratch wound assay
Marmoset astrocytes were plated on 8-well chamber slides coated with poly-D-lysine (2 × 104 cells/well) in DMEM supplemented with 10% FCS. After 24 h the medium was removed and the monolayer was scratched with a sterile 20–200 μL plastic pipette tip, as previously described (Goldshmit et al., 2004). The cells were washed three times with sterile PBS, and then 500 μL of growth medium was added, with or without cytokines. After 48 h in culture, the cells were fixed with 4% PFA for 10 min, followed by permeabilization with chilled (−20°C) methanol, and blocking with blocking solution (PBS containing 2% FCS and 2% GS) for 1 h. Immunostaining was performed using mouse anti-β-actin (1:10,000; Sigma-Aldrich), rabbit anti-EphA4 (1:300), and mouse anti-GFAP (1:1000; Invitrogen). The cells were incubated with primary antibodies for 2 h, and then chamber slides were washed three times in PBS. The cells were subsequently incubated with the secondary antibodies, goat anti-rabbit Alexa Fluor-488, and goat anti-mouse Alexa Fluor-594 (1:600 and 1:800, respectively; Molecular Probes, Eugene, OR) for 1 h at room temperature, washed three times in PBS, and mounted with Fluoromount (Dako North America). The nuclei of cells repopulating the scratched area were visualized by 4′-6-diamidino-2-phenylindole (DAPI; 1:1000; Molecular Probes) staining. The number of astrocytes entering the wound area was determined by counting the number of nuclei or GFAP-expressing cells per 0.035 mm2 in arbitrarily chosen sample areas along the border of the scratched area.
Results
Cellular changes proximal to the lesion site following short-term recovery
Three weeks following the V1 lesion, marmoset monkey cerebral tissues were processed and examined histologically. Nissl staining was used to demarcate the borders of the lesioned region of V1 (boxed region in Fig. 1B). Examination of the border of the lesion site for astrocytic gliosis revealed hypertrophic astrocytes immunostained very strongly for GFAP (Fig. 1C). In addition, an extensive reduction in neuronal cells was observed around the lesion site, as assessed by NeuN immunolabeling and DAPI staining (Fig. 1D). Apoptotic death of neuronal cells was examined by double immunolabeling with the neuronal marker NeuN and active caspases-3 expression (Fig. 1E). These results demonstrate neuronal loss in the borders of the ablated area, and the accumulation of reactive astrocytes creating the glial scar, indicative of an extensive cerebral injury. In the contralateral hemisphere the cortical tissue appeared normal; the astrocytes were not hypertrophic and GFAP expression was lower than in the lesioned hemisphere (Fig. 1G).
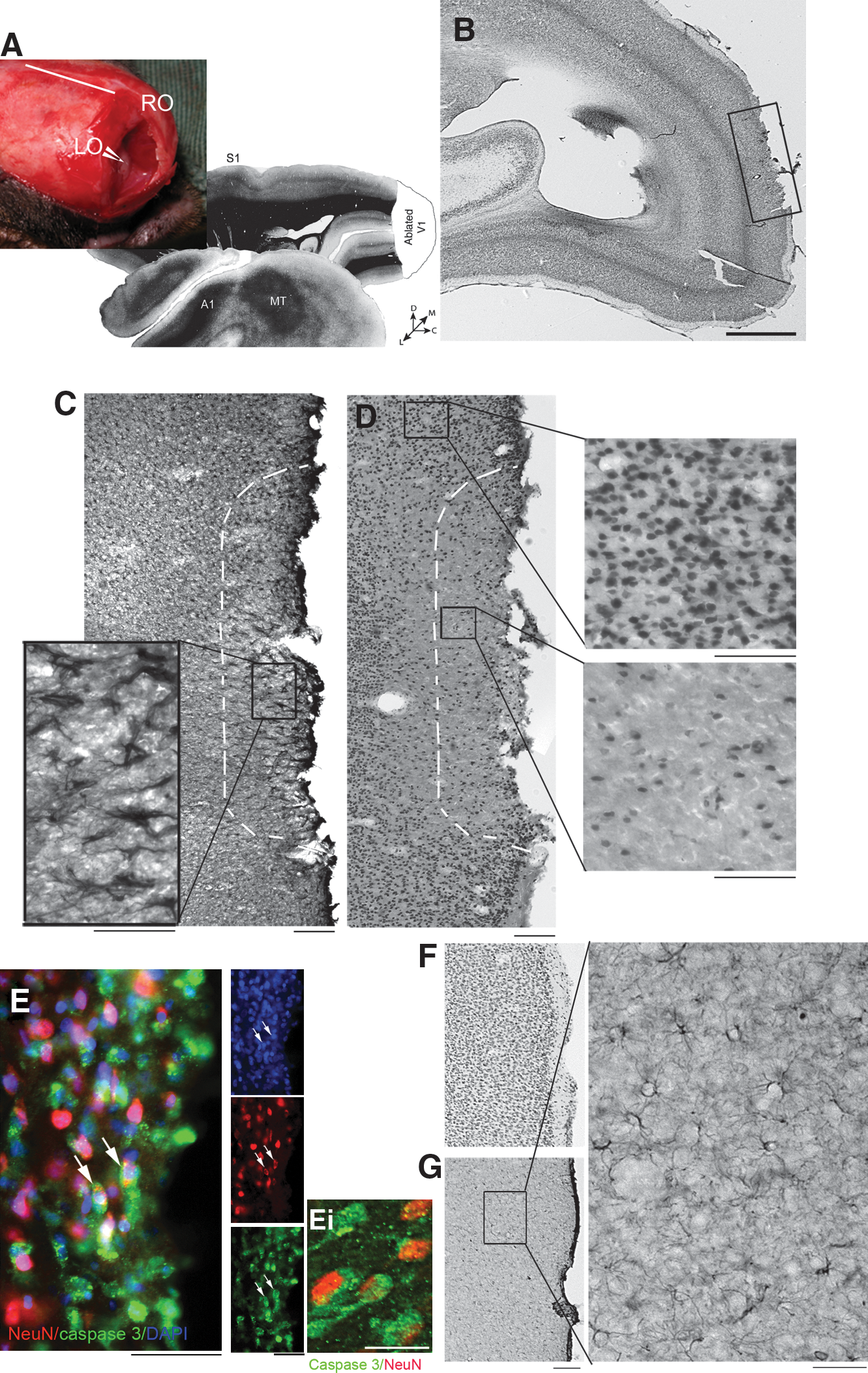
Cortical ablation leads to astrocytic gliosis and neuronal cell death at the border 3 weeks after lesioning. (
EphA4 expression is upregulated on astrocytes bordering the lesion site
Double immunolabeling with antibodies against GFAP and EphA4 revealed that EphA4 was strongly upregulated on the reactive astrocytes at the lesion site (Fig. 2A–C). Expression was upregulated on astrocytes in both the grey (Fig. 2A) and the white matter (Fig. 2B and C). The extent of EphA4 co-localization with astrocytes was examined in various regions of the lesioned adult brain; however, co-localization with the astrocyte marker GFAP was observed only at the lesion site (Fig. 2A and B). In astrocytes bordering the lesioned area, EphA4 immunolabeling was primarily confined to cell bodies and the major process branches. EphA4 expression was not examined on other glial cell populations, such as oligodendrocytes of microglia; however, the majority of the immunolabeling was specifically associated with astrocytes. In the contralateral intact V1, EphA4 immunolabeling was low in the white (Fig. 2D) and grey matter (Fig. 2F) of the cortex, and was predominantly localized to astrocytes around blood vessels (arrow in Fig. 2F).
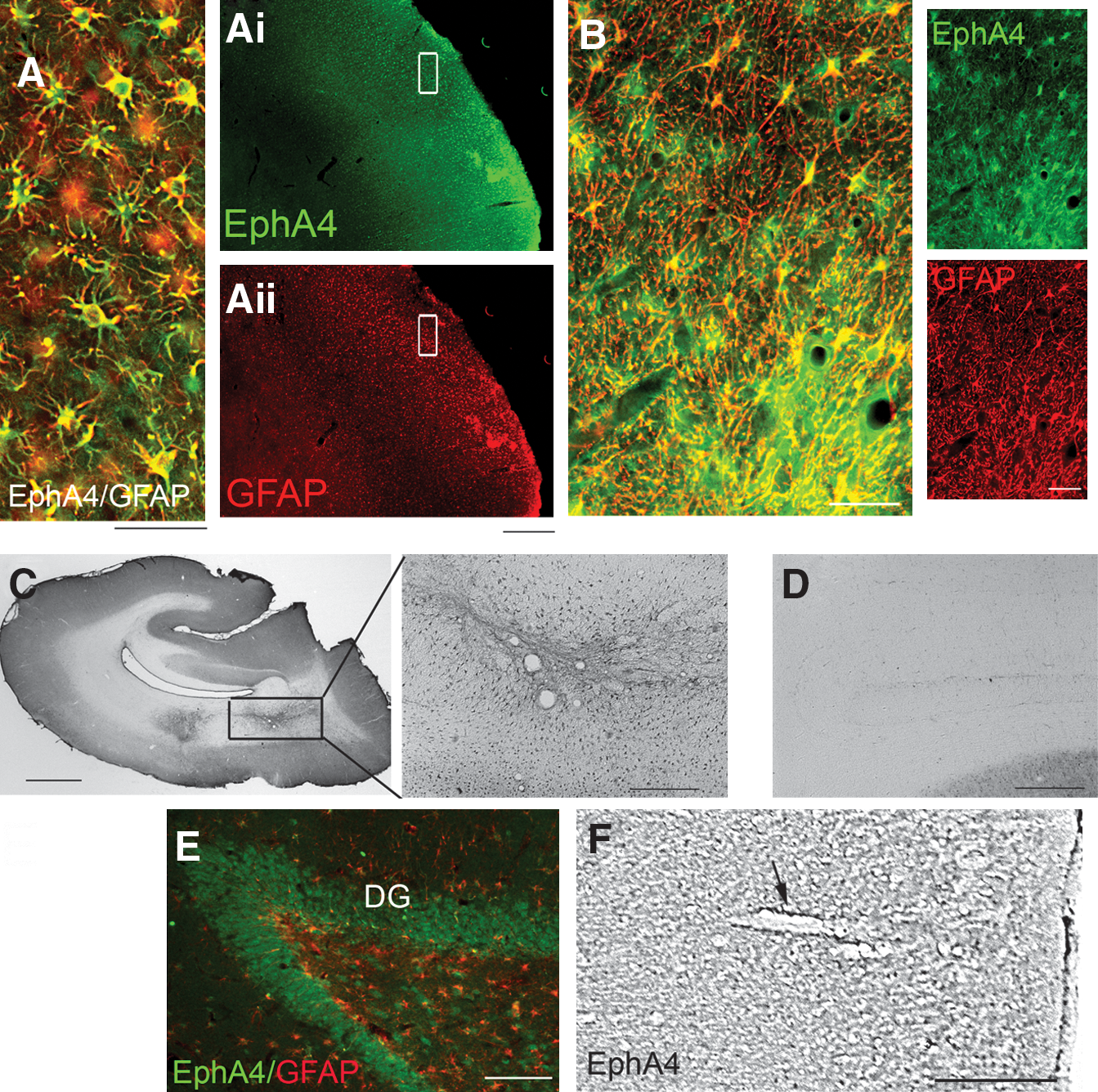
EphA4 is upregulated on reactive astrocytes only at the lesion site in the grey and white matter 3 weeks after lesioning. (
In the lesioned hemisphere, EphA4 expression was also observed distal to the lesion site, on neuronal cells in the dentate gyrus of the hippocampus (Fig. 2E), suggesting that EphA4 expression on hypertrophic and activated astrocytes is specific to the lesion site.
Chondroitin sulfate proteoglycans are secreted by EphA4-expressing astrocytes at the lesion site
Examination of CSPG secretion 3 weeks after cerebral injury in the marmoset revealed that CSPGs were secreted at the border of the lesion site (Fig. 3A). CPSG immunolabeling was concentrated at the injury site, and was diffuse throughout the extracellular matrix (ECM). Double immunostaining for GFAP and CPSG revealed that CSPG labeling was localized around the hypertrophic/reactive astrocytes in this area (Fig. 3B), which co-expressed high levels of EphA4 (Fig. 3C).
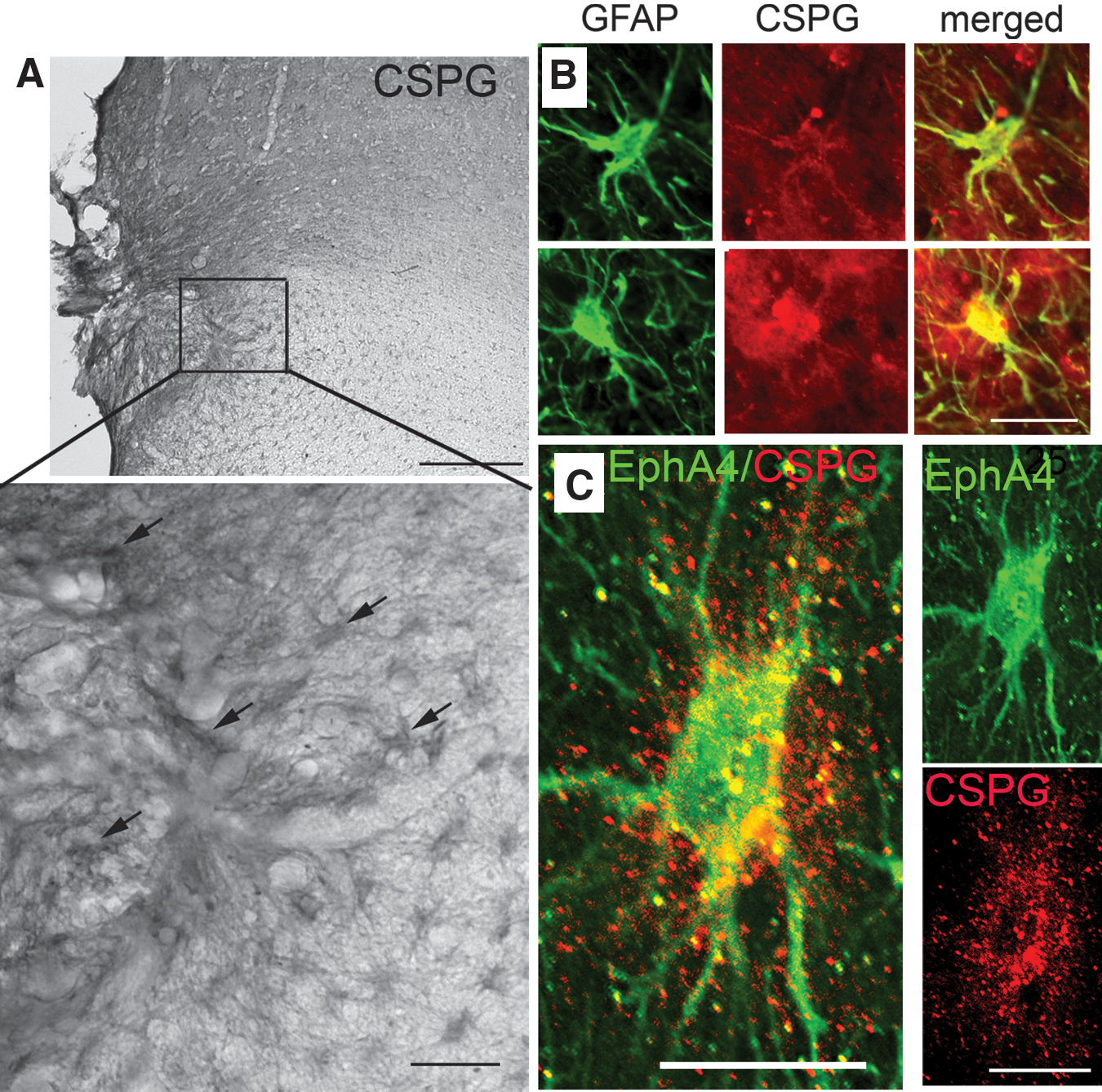
EphA4-expressing astrocytes increase expression of chondroitin sulfate proteoglycans (CSPGs) at the border of the lesion. (
Scratch wound repair is enhanced by EphA receptor activation
To determine whether glial scar formation is facilitated by EphA receptor activation in the injured marmoset brain, primary astrocyte cultures were established using the ablated cortical tissue. Cultured astrocytes were examined in vitro using the scratch wound assay. EphrinA5-Fc potentially binds with high-affinity to EphA4, and its other high-affinity partners EphA3, EphA5, EphA6, and EphA7 (Himanen et al., 2004). Clustered ephrinA5-Fc promotes EphA4 phosphorylation and downstream signaling in astrocytes and in neurons, inhibiting neurite outgrowth (Goldshmit et al., 2004; Wahl et al., 2000). LIF was used as a positive control for astrocyte proliferation (Goldshmit et al., 2004).
After 48 h, astrocyte migration into the scratched area was enhanced in the presence of clustered ephrinA5-Fc, compared with untreated astrocytes (Fig. 4A and B). The total number of astrocytes migrating into the scratched area significantly increased with both clustered ephrinA5-Fc and LIF treatment compared with untreated controls (Fig. 4B). In addition, the number of astrocytes that upregulated GFAP expression significantly increased in this area with clustered ephrinA5-Fc and LIF treatment (17% and 22%, respectively), compared to untreated astrocytes (4%; Fig. 4A, B, and C).
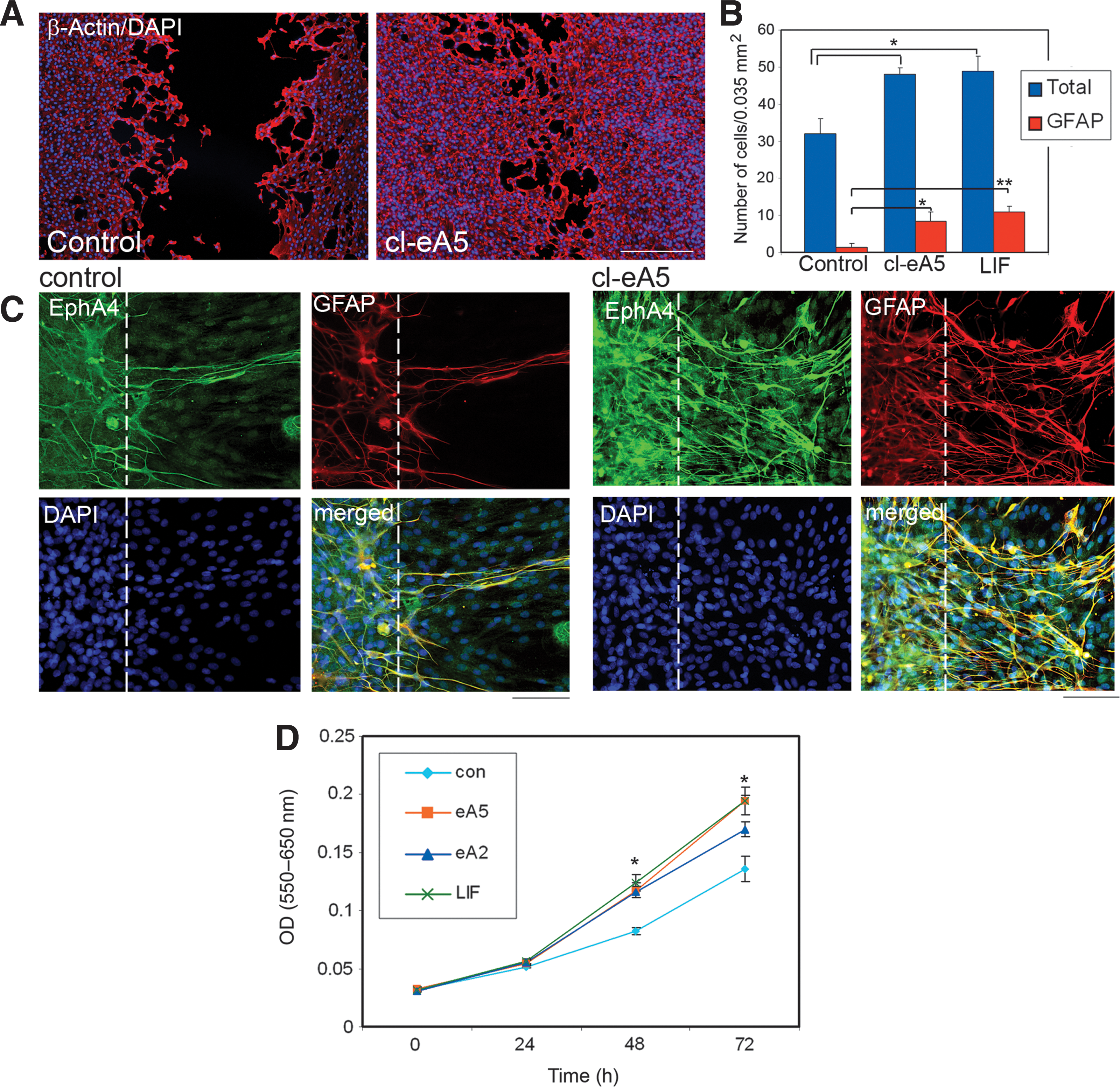
Repair of scratch wound and astrocyte proliferation in vitro promoted by EphA4 receptor activation. Primary marmoset astrocytes were derived from ablated V1 cortical tissue. Confluent astrocyte monolayers were scratched with a pipette tip and cultured in the presence or absence of clustered ephrinA5-Fc. Monolayers were fixed and stained for β-actin and DAPI, at various time points (DAPI, 4′-6-diamidino-2-phenylindole). (
Not all astrocytes in culture expressed high levels of GFAP; however, when an injury assay such as a scratch wound assay is used, many of the astrocytes around the scratched area upregulated GFAP. Interestingly, double immunostaining against GFAP and EphA4 revealed that these astrocytes also upregulated EphA4 expression (Fig. 4C), and that the number of these astrocytes was significantly higher when treated with clustered ephrin-A5. EphA4 was also detectable at low levels in cells with low or undetectable levels of GFAP expression in this assay.
Marmoset astrocytes proliferate in response to EphA4 receptor activation
Marmoset astrocyte proliferation was measured using the MTT proliferation assay. Both ephrinA5-Fc and ephrinA2-Fc show high-affinity binding to the EphA4 receptor, while ephrinA5-Fc shows higher-affinity binding than ephrinA2-Fc (Bowden et al., 2009). In the presence of the EphA4 receptor ligands clustered ephrinA5-Fc or clustered ephrinA2-Fc, astrocyte proliferation was significantly increased compared with untreated controls (Fig. 4D). After 72 h, EphA4 stimulation with clustered ephrinA5 or clustered ephrinA2 enhanced culture growth by 44% or 21%, respectively, compared with untreated astrocytes. Although clustered ephrinA5 led to higher percentage of proliferation than clustered ephrinA2, the difference was not significant. Furthermore, proliferation of astrocytes was similarly enhanced by the presence of LIF, a known inducer of astrocyte proliferation. This served as a positive control.
EphA4 receptor activates the Rho and mitogen-activated protein kinase pathways on marmoset astrocytes
In order to examine signaling through the EphA4 receptor in marmoset astrocytes, clustered ephrinA5-Fc was added to serum-starved cultures. At various time points, protein was harvested from the cells, subjected to immunoprecipitation against phosphorylated tyrosine residues, and analyzed by Western blot. Phosphorylated EphA4 receptor was virtually undetectable in serum-starved astrocyte cultures. However, 10 min after the addition of clustered ephrinA5-Fc, EphA4-receptor phosphorylation increased markedly, by 2.5-fold, with peak activation observed after 30 min (Fig. 5A). EphA4 receptors remained strongly activated after 2 h of continuous exposure to ephrinA5-Fc (Fig. 5A). The total concentration of EphA4 receptor remained the same in all samples, and did not change in the short term (up to 2 h). This was confirmed with a β-actin loading control (data not shown). Other EphA receptors, such as EphA3, EphA5, and EphA7, were examined for their expression in primate astrocytes and their ability to be activated by ephrinA5-Fc. The levels of these receptors were very low in astrocytes, and only EphA5 showed minimal activation with the clustered ephrinA5-Fc (data not shown).
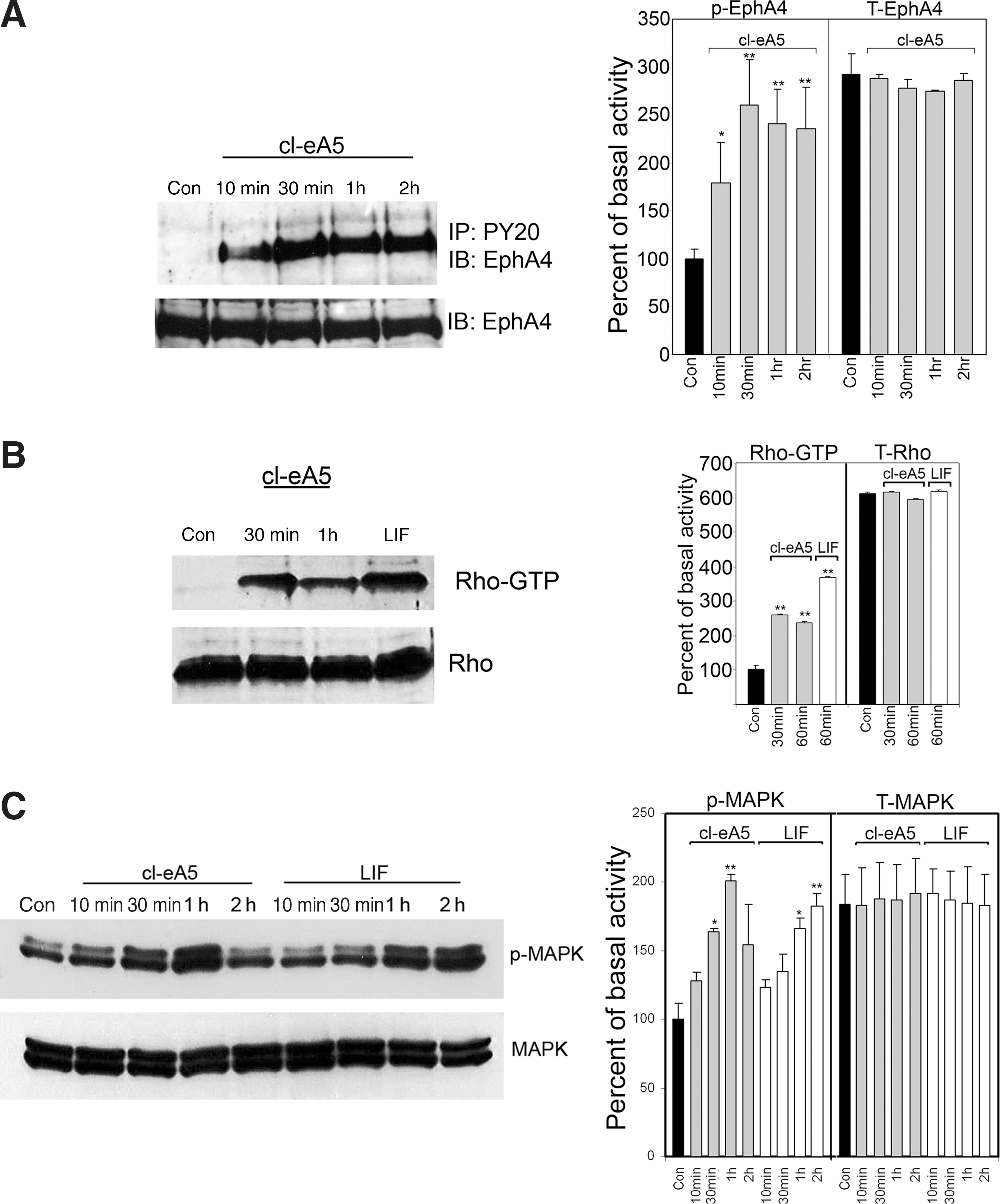
Ligand-induced activation of the mitogen-activated protein kinase (MAPK) and Rho pathways. Primary marmoset astrocyte cultures were serum-starved for 2 h (con) before the addition of either clustered ephrinA5-Fc (1.5 μg/mL) or LIF (1000 U/mL). Whole-cell lysates were prepared at 10, 30, 60, and 120 min. (
EphA4-receptor phosphorylation was correlated with activation of the downstream signaling effectors MAPK and RhoA. Active GTP-bound RhoA was undetectable in serum-starved astrocyte cultures; however, 30 min after the addition of clustered ephrinA5-Fc, a 2.5-fold increase in RhoA-GTP was detected by Western blot compared to basal levels in control, untreated cells (Fig. 5B). LIF activated RhoA in the marmoset astrocytes (Fig. 5B), in a similar temporal manner to that previously demonstrated in mouse astrocytes (Goldshmit et al., 2004). Total Rho levels were similar among all samples.
MAPK remained phosphorylated in astrocyte cultures in basal conditions, even after 2 h of serum starvation. After 10 min of clustered ephrinA5-Fc exposure, MAPK activation was unchanged, significantly increasing above basal levels after 30 min. Phosphorylated MAPK continued to accumulate with longer exposure times (up to 1 h; a twofold increase), but had returned to near basal levels by 2 h (Fig. 5C). In contrast, addition of LIF resulted in a delayed increase in MAPK activation compared with EphA stimulation, for which MAPK phosphorylation increased above basal levels only after 1 h of continuous exposure (Fig. 5C).
Taken together, these results indicate that the response of marmoset astrocytes to clustered ephrinA5-Fc includes a rapid and sustained activation of the EphA4 receptor, followed by a delayed activation of the MAPK and Rho pathways.
Discussion
To date, most studies investigating reactive gliosis and glial scar formation in vivo or in vitro have been conducted in rodent models. However, it is very important to determine whether the events pertaining to the astrocytic responses following neurotrauma are similar in the primate, which possesses more complex astrocytes than the rodent. In this study we examined the role of EphA4 in astrocytic gliosis in the marmoset monkey, which has previously been associated with glial scar formation in the rodent (Goldshmit et al., 2004). Following partial ablation of the primary visual cortex (V1), there was development of astrocytic gliosis, with increased GFAP expression and CPSG secretion by reactive/hypertrophic astrocytes at the border of the lesion site. Three weeks after injury, we observed a reduction in neuronal density at the borders of the lesion. Furthermore, many of the surviving neurons were undergoing apoptosis, indicating that neurodegeneration was continuing at this protracted time point.
In the contralateral hemisphere and in ipsilateral brain regions distal to the lesion site, EphA4 expression was restricted to perivascular astrocytes and neurons in the hippocampus, consistent with previous reports of EphA4 expression in intact primate (Xiao et al., 2006) and rodent (Goldshmit et al., 2006; Liebl et al., 2003; Martone et al., 1997; Moreno-Flores and Wandosell, 1999) brain. Proximal to the lesion site, EphA4 was strongly expressed by reactive/hypertrophic astrocytes in the grey and white matter. In contrast to unaffected brain regions, EphA4 expression was localized to astrocytes specifically expressing high levels of GFAP and secreting CPSGs into the ECM.
Following spinal cord injury of wild-type mice, EphA4 upregulation was observed in reactive astrocytes with increased GFAP expression and CPSG secretion. In contrast, EphA4-deficient mice were demonstrated to have significantly reduced GFAP expression and CSPG accumulation in the glial scar (Goldshmit et al., 2004). This reduced astrocyte reactivity and glial scarring in EphA4-knock-out mice facilitated neuronal regeneration, with regenerating axons penetrating through the scar. The data presented here extends these findings to include the primate brain, and provides evidence for the conservation of the role of EphA4 in astrocyte reactivity between primates and rodents.
In vitro analysis of primary marmoset astrocyte cultures using the scratch wound assay revealed that EphA4 receptor activation enhanced proliferation of primate astrocytes and enhanced closure in cultured monolayers. The astrocytes that migrated into the scratched area expressed high levels of GFAP and EphA4, suggesting a role for EphA4 signaling in primate astrocyte reactivity. Furthermore, these results are concomitant with those previously observed in the rodent (Goldshmit et al., 2004), providing further evidence of the conserved role of the EphA4 receptor.
We have demonstrated in the nonhuman primate that activation of Eph receptors by ephrins leads to a marked increase in the activation of Rho GTPases, which was previously observed in rodents (Dickson, 2001; Shamah et al., 2001; Wahl et al., 2000), and MAPK signaling (Carbonell and Mandell, 2003; Wang et al., 2005). Both the Rho- and MAPK-signaling pathways have previously been demonstrated to mediate the astrocytic response after injury (Brown and Bal-Price, 2003; Carbonell and Mandell, 2003; Dubreuil et al., 2003; Minghetti, 2005; Lim et al., 2007; Wang et al., 2005). MAPK pathway activation has also been associated with enhanced proliferation and migration of astrocytes derived from pluripotent embryonal human teratocarcinoma cells (Lim et al., 2007). Furthermore, we demonstrated that in the presence of the inflammatory cytokine LIF, that the Rho-signaling pathway was activated. In a previous study of EphA4-deficient astrocytes, activation of this signaling pathway was not observed (Goldshmit et al., 2004), suggesting that EphA4 signaling is necessary for the transduction of inflammatory signals in these cells.
A recent publication by Herrmann and colleagues (2010) suggests that EphA4-deficient mice do not demonstrate a significant reduction in astroglial-fibrotic scar formation after spinal cord injury, as was previously demonstrated by Goldshmit and colleagues (2004). However, we believe this difference may be due to the paradigm used in the quantification of GFAP-expressing astrocytes. Furthermore, a recent publication by Puschmann and Turnley (2010) showed that wild-type GFAP-expressing astrocytes had an increased proliferation rate after 24 h compared to astrocytes derived from EphA4-null mice. Their results suggest that stress fiber re-arrangement in EphA4-deficient astrocytes is decreased, and the adhesion of these cells is impaired; therefore, responses to stimuli that require cytoskeletal involvement are impaired in the EphA4-null astrocytes. These results may explain the prolonged blood–brain barrier permeability after spinal cord injury that was observed in the EphA4-deficient mice, for which GFAP expression on astrocytes around blood vessels was significantly reduced (Goldshmit et al., 2006). Moreover, in human malignant astrocytoma cells that overexpress EphA4, this molecule activation accelerated cell proliferation and migration through both MAPK and small-GTPases pathways (Fukai et al., 2008). These studies indicate that EphA4 may mediate astrocyte proliferation after injury or under other pathological conditions, and may contribute to scar formation.
Taken together, the observations made in this nonhuman primate study indicate a similar role for EphA4 in astrocyte reactivity that was previously uncovered in rodent models. Although it is still not clear whether expression and upregulation of EphA4 on astrocytes at the injury site regulates astrocytic gliosis, or whether inflammation increases EphA4 expression and activity, these studies provide further evidence that the upregulation and activation of EphA4 on astrocytes after brain trauma may be inhibitory for regeneration via multiple mechanisms. Therefore, blockade of the EphA4 receptor following brain trauma with, for example, unclustered EphrinA5-Fc, may be of therapeutic benefit and enable regeneration of severed neurons in a clinical setting.
Footnotes
Acknowledgments
The authors thank Prof. Marcello Rosa (Monash University) for providing brain tissue, which was collected as part of experiments funded by National Health and Medical Research Council (NHMRC) Project Grant 491022. Also, the authors would like to thank Dr. Samuel McLenachan for revision of earlier versions of this manuscript. This work was supported by the Australian Regenerative Medicine Institute; an NHMRC Project Grant 433620 (J.A.B.); a VNI Fellowship to Y.G.; and an R.D. Wright Fellowship (NHMRC) to J.A.B.
Author Disclosure Statement
No competing financial interests exist.