
Abstract
Chronic subdural hematoma (CSDH) is an angiogenic disease that is recognized as a cause of treatable dementia with unknown pathogenesis. Vascular endothelial growth factor (VEGF), a potent growth factor regulating angiogenesis through the phosphatidylinositol 3-kinase (PI3-kinase)/Akt pathway, has been implicated in its etiology. The status of this signaling pathway in CSDH outer membranes was examined in the present study, using outer membranes obtained during trepanation surgery. Expressions of PI3-kinase, PKB-kinase, Akt, phosphorylated Akt at Ser473 (p-Akt), endothelial nitric oxide synthase (eNOS), vascular endothelial-cadherin (VE-cadherin), and actin were examined by Western blot analysis, together with their immunohistochemistry. PI3-kinase, Akt, eNOS, and VE-cadherin were detected in all cases. The magnitude of the expression of p-Akt varied among cases; however, the localization was revealed to be present in endothelial cells of vessels in CSDH outer membranes, together with VEGF and VE-cadherin detected in endothelial cells of vessels. These findings suggest that the PI3-kinase/Akt signaling is activated in CSDH outer membranes, and indicate the possibility that the PI3 kinase/Akt pathway might be activated by VEGF and play a critical role in the angiogenesis of CSDH.
Introduction
C
Methods
Patients
Ten patients (8 men, 2 women; age 58–79 years; mean age 67 years) suffering from CSDH confirmed by computed tomography (CT) or magnetic resonance imaging (MRI) were enrolled in this study. All underwent burr hole irrigation surgery under local anesthesia at Aichi Medical University Hospital. All had a history of head injury and none had any hemostatic disorder or was receiving antiplatelet or anticoagulation therapy. The clinical data are presented in Table 1.
The size refers to the largest extent measured in pre-operative CT slices. Shift refers to the midline shift.
M, male; F, female; DWI, diffusion-weighted imaging; CT, computed tomography; FLAIR, fluid attenuated inversion recovery.
Western blot analysis
Samples of outer membranes from CSDH were obtained during trepanation surgery, immediately homogenized with Laemmli sample buffer, and centrifuged at 12,000g for 10 min at 4°C. Supernatant samples were subjected to 7.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, and proteins were then transferred to polyvinylidene difluoride (PVDF) membranes and incubated with primary polyclonal antibodies against actin (Sigma-Aldrich, St. Louis, MO), endothelial nitric oxide synthase (eNOS; BD Transduction Lab, Franklin Lakes, NJ), vascular endothelial-cadherin (VE-cadherin; Cell Signaling Technology, Danvers, MA), and phosphorylated Akt at Ser473 (p-Akt; Cell Signaling Technology), and primary monoclonal antibodies against phosphatidylinositol 3-kinase (PI3-kinase; BD Transduction Lab), and PKB kinase (BD Transduction Lab), at a dilution of 1:500 overnight at 4°C. After washing, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (Sigma-Aldrich) at a dilution of 1:3000 for 30 min at room temperature. Reactions were developed with ECL or ECL plus (GE Healthcare, Buckinghamshire, U.K.). Phosphorylated Akt at Ser473 immunoblots were stripped from PVDF membranes and reblotted with primary monoclonal Akt (BD Transduction Lab) at a dilution of 1:500 overnight at 4°C. Finally, the membranes were developed with ECL plus.
Histological examination and immunofluorescence staining
To study the cellular localization of activated Akt, VEGF, and VE-cadherin, double staining of Akt/von Willebrand factor (vWF), p-Akt/vWF, VEGF/vWF, and VE-cadherin/vWF was performed (n=3). Outer membranes were prepared according to the methods described below. Samples were preserved in 10 mL of ice-cold 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for 3 h and rinsed with 0.1 mol/L lysine hydrochloride in 0.1 mol/L phosphate-buffered saline for 3 h. Serial axial cryostat sections (10 μm) were collected on silane-coated slides for staining. At first, these sections were stained with hematoxylin and eosin (H&E; n=3). Next, non-specific immunoreactivity was blocked with fetal bovine serum. After membrane permeabilization in 0.1% Triton X-100, 100 mmol/L sodium phosphate (pH 7.4), and 150 mmol/L sodium chloride, the samples were treated with primary antibodies against Akt, p-Akt, or VE-cadherin (Cell Signaling Technology) at a dilution of 1:100, VEGF (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at a dilution of 1:50, and vWF (mouse monoclonal antibody; Dako, Glostrup, Denmark) at a dilution of 1:100 overnight at 4°C. Primary antibody label of Akt, p-Akt, VE-cadherin, and VEGF was detected with an Alexa Fluor 488-labeled IgG secondary antibody (Invitrogen, Inc., Eugene, OR) at a dilution of 1:200. The vWF primary antibody label was with Alexa Fluor 546-labeled IgG secondary antibody (Invitrogen) at a dilution of 1:200. To demonstrate the specificity of the primary antibodies, controls of immunohistochemistry are presented with the non-immune IgG fraction of the rabbit serum. Some of the specimens were stained with 0.5 μg/mL 4′,6-diamino-2-phenylindole (DAPI) (Invitrogen) in 20 mmol/L sodium phosphate (pH 7.4) and 150 mmol/L sodium chloride for 5 min for visualizing nuclei. The samples were then examined with an Apo Tome (Carl Zeiss Co. Ltd., Jena, Germany) and a differential interference contrast (DIC) microscope or a confocal laser scanning microscope (Carl Zeiss).
Results
Western blot analysis of Akt, p-Akt, PI3-kinase, PKB-kinase, eNOS, and VE-cadherin
The degree of expression of p-Akt differed in each case (Fig. 1). Nearly constant levels of actin were detected in all cases, suggesting essentially equal levels of protein applied to the SDS gels. PI3 kinase, PKB kinase, Akt, eNOS, and VE-cadherin were also detected in all cases. PKB kinase, however, was weaker in some cases.
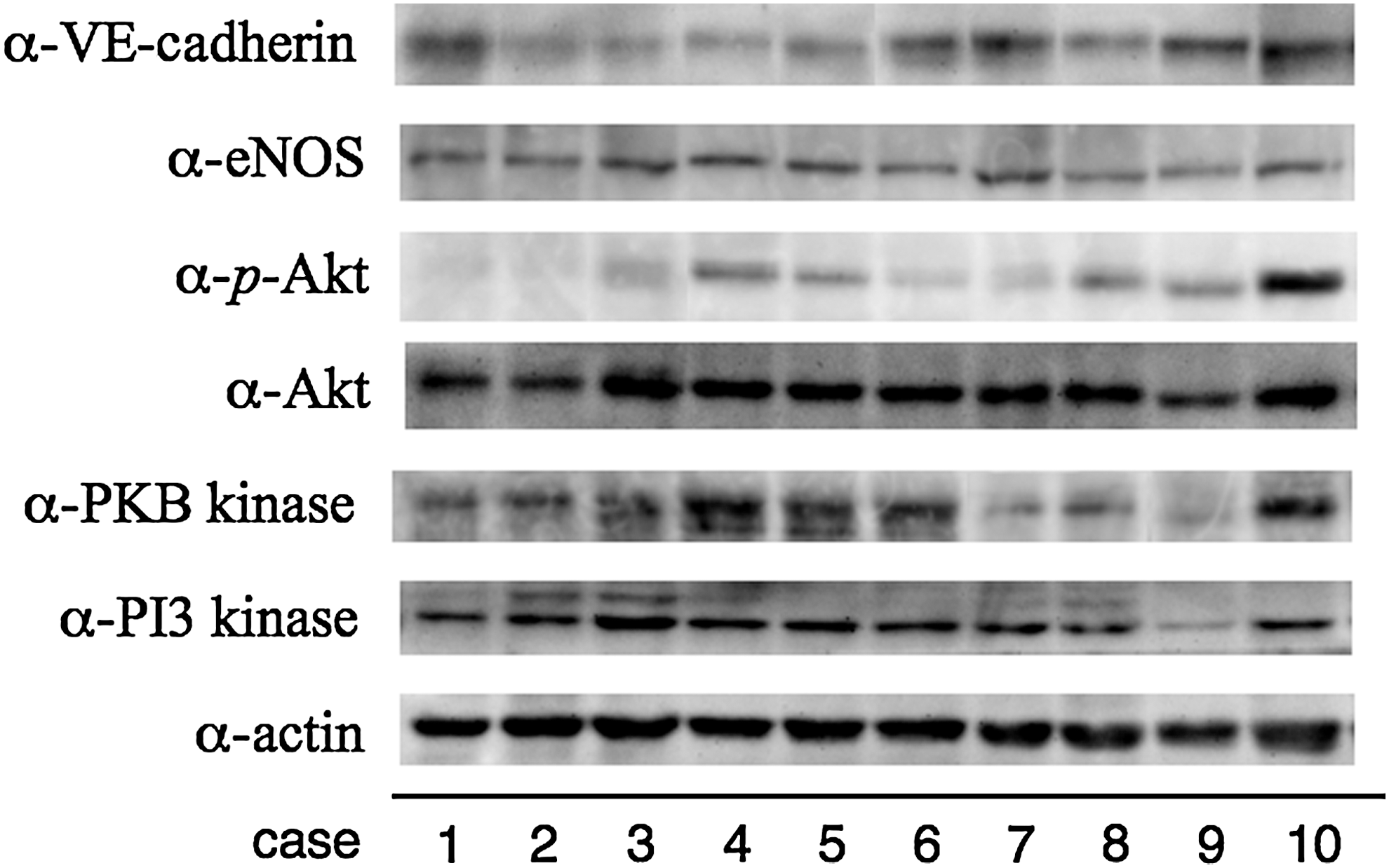
Outer membranes of chronic subdural hematomas from 10 patients homogenized with Laemmli sample buffer were subjected to Western blotting with anti-actin (α-actin), anti-phosphatidylinositol 3 kinase (α-PI3 kinase), anti-PKB kinase (α-PKB kinse), anti-Akt (α-Akt), anti-phosphorylated Akt at Ser473 (α-p-Akt), anti-endothelial nitric oxide synthase (α-eNOS), and anti-vascular endothelial-cadherin (α-VE-cadherin) antibodies. Note that almost all molecules except p-Akt could be detected in outer membranes of chronic subdural hematomas. Activation of Akt (p-Akt/Akt) varied with the case.
Histological observations
H&E staining showed that vessels were well developed and inflammatory cells including eosinophils were observed between fibroblasts (Fig. 2A). vWF is a large multimetric glycoprotein present in blood plasma which is produced constitutively in endothelium. Immunofluorescence staining of vWF together with differential interference contrast observation revealed that vWF was positive, mainly in the endothelium of vessels (Fig. 2B). Confocal immunofluorescence analysis of vessels, using double staining for Akt (green), or activated Akt (green) and vWF (red), showed that endothelial cells of vessels in the outer membrane were immunopositive for Akt (Fig. 2C) and activated Akt (Fig. 2D and E). VEGF and VE-cadherin were also detected in the endothelium of vessels (Fig. 2F and G, respectively). Immunofluorescence stainings of chronic subdural membranes with the IgG fraction of non-immune rabbit serum were consistently negative (data not shown).
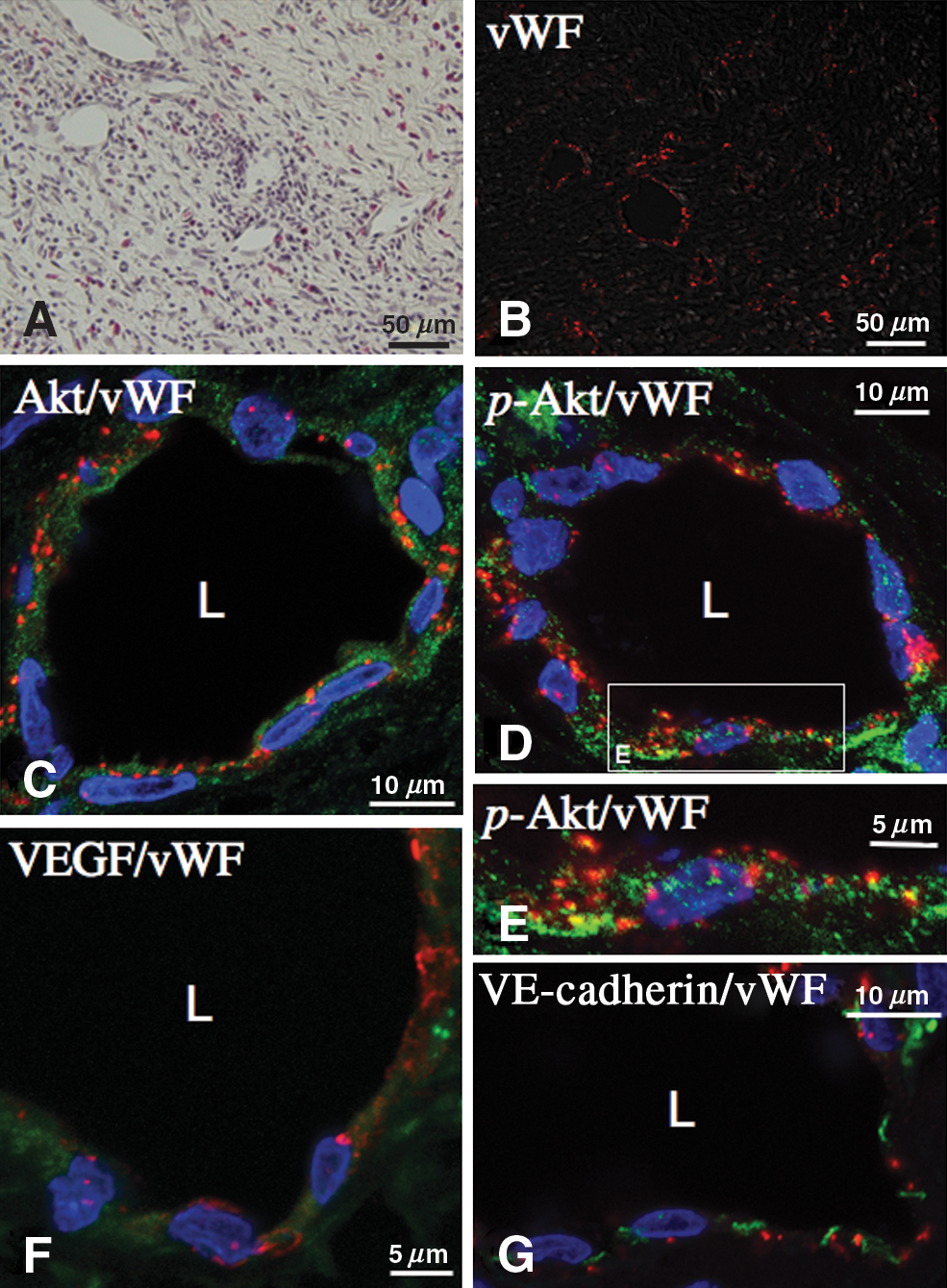
(
Discussion
In this study we clarified that VEGF, PI3 kinase, PKB kinase, and Akt were expressed in CSDH outer membranes along with activation of Akt in the endothelial cells of vessels.
CSDH is considered to be an inflammatory and an angiogenic disease. Inflammatory cytokines like interleukin (IL)-6 and IL-8 play a role in its development and are significantly increased in hematoma fluid compared to serum (Suzuki et al., 1998). IL-6 can induce enlargement of the gap junction of endothelial cells and increase vascular permeability by rearranging actin filaments and by changing the shape of endothelial cells (Maruo et al., 1992). A previous study using electron microscopy revealed that endothelial gap junctions were widely open and played an important role in the leakage of blood, causing enlargement of CSDH (Yamashima et al., 1983). IL-8, which is a cytokine that is chemotactic for lymphocytes and neutrophils, is also a potent angiogenic factor (Koch et al., 1992). Prospective studies have revealed that high levels of both IL-6 and IL-8 are significantly associated with recurrence (Frati et al., 2004; Hong et al., 2009). Excessive activation of fibrinolytic systems is also considered to be a possible etiological factor for the development of CSDH. Tissue plasminogen activator (t-PA), one of the coagulofibrinolytic factors, has been revealed to be involved with the induction of intermittent bleeding by conversion of plasminogen to plasmin from the CSDH outer membranes (Ito et al., 1988). Patients with high t-PA concentrations in hematoma fluid have a relatively high probability of recurrence (Katano et al., 2006). VEGF increases t-PA activity in a dose-dependent manner in endothelial cells (Pepper et al., 1991), and is a potent vascular endothelial cell mitogen that induces angiogenesis. The significant angiogenic effects of VEGF are observed in both developing and adult brains (Rosenstein et al., 1998), especially in the ischemic brain, improving neurological recovery (Zhang et al., 2000). VE-cadherin is a strictly endothelial-specific adhesion molecule expressed at interendothelial junctions of tumor vessels (Vestweber, 2008), which is in agreement with our immunofluoresence results. VE-cadherin is essential during embryonic angiogenesis (Carmeliet et al., 1999), and is also activated in tumor-induced angiogenesis (Prandini et al., 2005). Previous studies have revealed that phosphorylation of VE-cadherin controls vascular permeability and angiogenesis (Potter et al., 2005; Hatanaka et al., 2011; Wallez et al., 2007). Whether VE-cadherin is phosphorylated in CSDH outer membranes awaits further study; nevertheless, these data suggest that VE-cadherin may be closely involved in angiogenesis in CSDH by VEGF.
VEGF has been reported to induce PI3 kinase activity in various endothelial cells, and serine/threonine protein kinase Akt is one of the major targets of PI3-kinase through PKB kinase. Furthermore, VEGF regulates endothelial cell survival through the PI3-kinase/Akt pathway (Gerber et al., 1998). Not only cell survival but also cell proliferation, permeability, release of nitric oxide, and cell migration are regulated by phosphorylation of Akt in endothelial cells. A recent report revealed that phosphorylation of girdin by Akt promotes VEGF-dependent migration of endothelial cells and tube formation (Kitamura et al., 2008). Our data suggest that VEGF may transduce its signals through PI3-kinase, PKB kinase, and Akt molecules in CSDH outer membranes.
Akt directly phosphorylates eNOS on serine 1179 and activates the enzyme, leading to NO production (Fulton et al., 1999). VEGF is also known to increase NO release in rabbit and human endothelial cells and to stimulate the growth of human umbilical vein endothelial cells in an NO-dependent manner, this being partially blocked by a PI3-kinase inhibitor (Papapetropoulos et al., 1997). NO plays a critical role in angiogenesis (Cooke and Losordo, 2002) and Matsunaga and associates (2002) have provided evidence that NO might suppress production of angiostatin, an endogenous antagonist of angiogenesis. Our finding that activated Akt is located in endothelial cells of CSDH outer membranes suggests that NO might be involved with angiogenesis in this disease.
Not only angiogenesis but also fibroblastic proliferation play important roles in the growth of CSDH. VEGF can induce the expression of connective tissue growth factor (CTGF), a well-known growth factor with actions on fibroblast proliferation, by selective activation of the PI3-kinase/Akt pathway (Suzuma et al., 2000). Expression of VEGF increases in response to myocardial ischemia and promotes vascular repair. Recently, VEGF in an infarcted heart was reported to mediate migration of cardiac stem cells (CSCs) to the ischemic area via activation of PI3 kinase/Akt (Tang et al., 2009), which is significantly attenuated by inhibition of phosphorylated Akt activity. These data suggest that the PI3 kinase/Akt pathway makes essential contributions to cell growth and repair. The growth of CSDH might depend on activated Akt through PI3 kinase/Akt signaling.
In summary, for the first time the present investigation has clarified the expression of PI3-kinase, PKB kinase, Akt, and VE-cadherin molecules in CSDH outer membranes. Of particular note is our finding that activation of Akt occurs in the endothelium of the vessels, suggesting that these molecules may be involved in angiogenesis in CSDH. However, we have to keep in mind that these angiogenic markers may represent an endogenous repair after injury rather than being a direct cause of disease per se. Further studies using inhibitors of this signaling pathway might more precisely clarify the mechanisms of growth of CSDH, and yield clinical applications to avoid re-growth of CSDH.
Footnotes
Acknowledgments
This work was supported in part by grants from the Smoking Research Foundation (Y.W.) and the Japan-China Medical Association (Y.W.), and also by a grant-in-aid for Scientific Research and High Technology Research Centre Project (19-8) (Y.W.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Dr. Malcolm Moore for critical reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.