
Abstract
The mechanisms by which Dilantin confers anticonvulsant benefits may also be neuroprotective by attenuating the acute excitatory insult in cortical and subcortical structures when the drug is given in the acute phase after traumatic brain injury (TBI). However, when Dilantin is used for prolonged periods, we hypothesized that it may impede recovery, synaptic plasticity may be impaired, and neuroprotective benefits may be lost. As such, we assessed the effect of daily chronic administration (75mg/kg day 0 followed by 50mg/kg daily i.p.) and acute administration (75mg/kg day 0 followed by 50mg/kg i.p. day 1) of Dilantin in young adult male rats on motor performance, y-maze exploration, Morris Water Maze (MWM), hippocampal (HC) cell survival, contusion size, and regional expression of neuroplasticity markers after controlled cortical impact (CCI) injury. Chronic daily Dilantin administration resulted in beam walking impairments on day 6, whereas acute Dilantin administration resulted in beam walking impairments on days 3 and 4. Chronic Dilantin administration also resulted in worse MWM performance, more HC cell loss and no increases in neuroplasticity markers compared to rats with CCI receiving chronic vehicle. Conversely, rats receiving acute Dilantin administration exhibited more novel arm exploration in the y-maze, greater HC cell sparing, and greater growth-associated protein 43 (GAP-43) expression in the HC ipsilateral to the CCI, compared to injured rats receiving vehicle. MWM was not influenced by acute Dilantin administration. These results suggest that there are beneficial effects of limited acute Dilantin therapy after TBI, and that extended daily Dilantin therapy has deleterious effects on neural recovery. These findings support clinical guidelines for limited use of Dilantin in seizure prophylaxis after TBI.
Introduction
T
However, the efficacy of Dilantin administration beyond the acute post-injury period is questionable, based on randomized controlled clinical trials (RCT) (Temkin et al., 1990). Long- term administration of Dilantin provides little benefit in preventing late onset seizures (beyond one week), and it can impair cognitive and motor function. In fact, Dikmen and colleagues (1991) noted significantly worse neurobehavioral deficits one month post-TBI in patients treated with Dilantin and tested in multiple areas of neuropsychological performance. Pullianen and Jokelainen (1995) also noted that poor performance with neuropsychological assessment in patients with epilepsy may be secondary to poor visually guided motor function implying possible effects of Dilantin on cerebellar function as well. The negative effects of prolonged administration of Dilantin potentially may be explained by its effects on the same neurotransmitters that provide the potential neuroprotection early after injury. During the post- acute phase of TBI, excitatory neurotransmitter systems, required for synaptic plasticity and recovery of function, are often depressed (Arckens et al., 2000). Whereas glutamate, when released in large concentrations, is known to lead to cell death, excitatory input is also required for the changes in neuron cyto-architecture associated with neural recovery (Mattson and Katter, 1989). Therefore, Dilantin may diminish excitotoxicity to confer neuroprotection during the acute injury phase, but ongoing use may impair synaptic plasticity and recovery of function.
A common clinical dilemma often faced by rehabilitation physicians admitting patients with TBI for acute rehabilitation is that there is inconsistency in clinical practice in PTS prophylaxis. The Association of Academic Neurologists (AAN) currently recommends early intervention with Dilantin (i.v.) given as a loading dose as soon as possible followed by a 7-day course in the asymptomatic moderate-to-severely brain injured population (Chang and Lowenstein, 2003). Despite these recommendations, physicians admitting TBI patients into acute rehabilitation programs encounter a significant portion of their population who have had Dilantin prescribed as prophylaxis for periods substantially longer than 1 week, whose serum Dilantin level is subtherapeutic, or who have received Dilantin even though they belonged to the subpopulation with mild TBI.
As such, we used the controlled cortical impact (CCI) model of TBI to explore the potential effects of acute and chronic administration of Dilantin on neural recovery with moderate–severe TBI. We used an acute (within the first 24 h) and chronic (daily) dosing regimen to study the effects of Dilantin following experimental TBI. Our experimental end- points included tests of motor function, spatial learning, HC cell survival, cortical contusion volume, and HC and cortical markers of synaptic plasticity. We hypothesized that acute dosing would be neuroprotective, whereas chronic dosing would have detrimental effects on motor function, spatial learning, and neural recovery.
Methods
Animals
All experimental procedures for this study were approved by the University of Pittsburgh's Institutional Animal Care and Use Committee, in accordance with the standards of the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. Adult male Sprague–Dawley rats (Hilltop, Scottsdale, PA) were housed in standard steel wire mesh cages with a 12 h on/12 h off light/dark cycle, constant temperature (21 ± 1°C), and free access to food and water. Animals were housed in pairs and randomly assigned to receive CCI or sham surgery. All behavioral and histological procedures were conducted by assessors blinded to injury group/treatments, and exploratory behavioral assessments were videotaped for analysis. Two independent experimental studies were implemented to address the objectives outlined. Experiment 1 examined the effects of daily chronic Dilantin dosing on motor function, spatial learning, and neural recovery, whereas Experiment 2 examined the effects of acute Dilantin administration on motor function, spatial learning, and neural recovery.
Drug administration
Experiment 1
Rats were randomly assigned to an injury vehicle group (TBI + CV) (n = 15), an injury drug group (TBI + CD) (n = 15), a sham surgery vehicle group (SHAM + CV) (n = 12), or a sham surgery drug group (SHAM + CD) (n = 12). Mean weights at the time of injury were: TBI + CV, 325 ± 9.48 g; TBI + CD, 319 ± 8.33 g; SHAM + CV, 316 ± 2.99 g; and SHAM + CD, 318 ± 2.82 g. The chronic administration group received a 75 mg/kg injection of either drug or saline 15 min post-injury, and continued to receive injections every 24 h for the duration of the 21-day study. Injections were performed after daily testing, giving approximately 24 h between the injection and the next day of testing. The dosing paradigm was developed based upon previously described pharmacokinetics of Dilantin, to ensure that plasma levels were in the therapeutic but not the toxic range (Rundfeldt and Loscher, 1993). In addition, trunk blood was collected for analysis of serum drug levels from the SHAM + CD (n = 6) and SHAM + CV (n = 6) groups at the time of euthanasia (24 h after the final injection).
Experiment 2
Rats were randomly assigned to an injury vehicle group (TBI + AV) (n = 15), an injury drug group (TBI + AD) (n = 15), a sham surgery vehicle group (SHAM + AV) (n = 12), or a sham surgery drug group (SHAM + AD) (n = 12). Mean weights at the time of injury were: TBI + AV, 318 ± 3.78 g; TBI + AD 314 ± 4.86 g; SHAM + AV, 325 ± 2.69 g, and SHAM + AD 320 ± 3.39 g. The acute dosing group received a 75 mg/kg injection of either drug or saline 15 min post-injury, and a second 50 mg/kg injection 24 h post-surgery. The dosing paradigm was developed based upon previously described pharmacokinetics of Dilantin (Rundfeldt and Loscher, 1993).
CCI
The CCI injury device, described in detail by Dixon and colleagues (1991), consists of a small (1.975 cm) bore, double-acting stroke-constrained pneumatic cylinder with a 5.0 cm stroke. The cylinder is rigidly mounted on a crossbar in an angled position. The lower rod end has an impact tip with a diameter of 6 mm, and the upper rod end is attached to the transducer core of a linear velocity displacement transducer (LVDT; Schaevitz Model 500 HR, Macrosensors, Howard A. Schaevitz Inc., Pennsauken, NJ). The impact tip is pneumatically driven at a pre-programmed velocity, depth, and duration of tissue deformation. The velocity of the cylinder is controlled by gas pressure and measured directly by the LVDT.
Animals received induction anesthesia with inhalation of 4% isoflurane and a 2:1 N2/O2 mixture followed by 1–1.5% isoflurane for maintenance anesthesia. While under anesthesia, each animal was secured in a stereotactic frame. After midline incision and reflection of the soft tissues, a craniectomy (∼6 mm) was made between bregma and lambda in the right hemisphere between the central suture and the coronal ridge. The cortical injury was then delivered at approximately an 18 degree angle, such that the impactor was perpendicular with the dural plane. The impact was delivered to a depth of 2.9 mm at 4.0 m/sec. Core body temperature was monitored via rectal probe, and a homeothermic blanket (Harvard Apparatus, Hollison, MA) was used to maintain core body temperature at 37.0 ± 0.5°C. Animals receiving sham surgery underwent the same procedures with the exception of the injury.
Following surgery, recovery of consciousness was assessed using time until restoration of tail pinch, paw pinch, and righting reflexes. There were no significant differences across groups in either experiment (data not shown).
Motor performance
To assess motor deficits after surgery, both beam balance and beam walking tests were employed, as previously described, using assessors blinded to the treatment (Wagner et al., 2004). Beam balance testing consisted of measuring the duration (60 sec maximum) of time that rats balanced on a narrow (1.5 cm) beam elevated (90 cm) above the ground. For the beam walking task, the latency (60 sec maximum) to traverse a 100-cm beam and reach a goal box when presented with an adverse stimulus (white noise) was measured. Termination of the white noise upon entering the darkened goal box served as the reward for completing the task. If the rat fell off of the beam prior to reaching the goal box, it was assigned a latency of 60 sec. The beam (including goal box) was divided into equal zones labeled 1–5, such that a score of 1–5 was assigned based upon which zone (if any) rats fell from. A score of 5 was assigned if rats successfully reached the goal box. Rats were trained on beam walking 1 day prior to surgery, and pre-assessed for both tasks on the day of surgery to verify that rats could perform each task. For days 1–6 post-surgery, rats were evaluated on both tasks by completing three trials each testing day.
Y-maze testing
One y-maze testing session was conducted for each rat on day 7 post-surgery, using assessors blinded to the treatment. The task was adapted from Zhuang and colleagues (2001) for use with rats. The y-maze apparatus had three arms (34 × 10 × 18 cm) set at a 120 degree angle from each other and was equipped for video monitoring–recording. Each of the three arms contained a unique textured material: sandpaper, bubble wrap, or blue contact paper placed on the floor and walls. Preliminary testing was done to ensure that there was no exploratory preference for any of the textures (data not shown). To begin habituation, one of the three arms was partitioned off. Each rat was then placed in the center of the testing device and allowed to explore the other two arms for 10 min. Following the 10-min habituation period, the partition was removed, and each rat was allowed to explore all three arms for 5 min. The total time spent in each of the arms was recorded. The amount of time in the novel arm was compared to the combined time spent in the familiar arms divided by a factor of two. Time spent in the middle of the apparatus, without the full head and forepaws being in an arm, was not included in the analysis. The apparatus was cleaned with disinfectant after each session, and the textured materials were replaced routinely.
Morris Water Maze
The Morris water maze (MWM) (Morris, 1984), a task previously demonstrated to be sensitive to cognitive function/dysfunction (Dixon et al., 1999 a,b; Hamm, 2001; Hamm et al., 1992 a,b; Kline et al., 2000; Scheff et al., 1997), was used to measure spatial learning ability. The MWM apparatus consisted of a large plastic tub (128 cm diameter, 60 cm high) in a testing room containing visual cues that remained constant throughout the study. A clear Plexiglas platform (10 cm diameter, 26 cm high) was placed 26 cm from the wall of the tub in the southwest quadrant. The tub was filled with tap water (26 ± 1°C) to a depth of 28 cm, submerging the escape platform and rendering it invisible to the rat. Spatial learning acquisition testing was performed on days 14–18 post-surgery, using assessors blinded to the treatment. Each session of testing consisted of four trials with a 4 min inter-trial interval. Each of the four trials began with the rat facing the wall of the tub at one of the four cardinal directions (north, south, east, west). The trials were randomly assigned so that the rat started at all four of the directions in a testing day. Each trial lasted until the rat successfully mounted the platform, or a maximum of 120 sec had elapsed. If the rat failed to locate the platform in the allotted time, it was guided to the platform. Each rat remained on the platform for 30 sec before being placed in a heated incubator until the next trial. The times to locate the platform and swimming distance were recorded for each group. On day 19 post-injury, probe trials were performed by removing the hidden platform and allowing the rat 30 sec to swim in the pool. For analysis, the pool space was divided into three concentric circles, with the middle circle containing the escape platform. The time spent in the concentric circle containing the platform was recorded and averaged for each group. Swim speeds were also measured to assess motor function during acquisition and probe sessions.
Quantification of HC neurons
Three weeks after CCI or sham injury, animals [TBI + CV (n = 7), TBI + CD (n = 8), TBI + AV (n = 6), TBI + AD (n = 6)] were perfused transcardially while under deep anesthesia with pentobarbital (50 mg/kg, i.p.) using 200 mL heparinized 0.1 M phosphate-buffered saline (pH 7.4) and 300 mL 10% buffered formalin. The brains were extracted, post-fixed in 10% buffered formalin for 1 week, dehydrated with alcohols, and embedded in paraffin. Seven-micrometer thick coronal sections were cut at 1 mm intervals through the contusion on a rotary microtome and mounted on glass microscope slides. After drying overnight, the sections were deparaffinized in xylenes, rehydrated, and stained with Cresyl violet. One coronal section underlying the area of contusion (∼3.5mm posterior to bregma) from each rat in all groups was analyzed for determination of treatment effects on HC CA1 and CA3 neurons. To reduce counting errors associated with false-positive identification of dying neurons, the total number of CA1 and CA3 morphologically intact neurons (i.e., those with a clearly defined cell body and nucleus) were counted using a Nikon Eclipse E600 microscope (Nikon Corporation, Tokyo, Japan) with a 40x objective. Neurons that were partially observed because of sectioning were not included. All data were reported as the percent of total neurons in the ipsilateral (injured) CA1 and CA3 regions relative to each contralateral HC region.
Cortical lesion volume
The area of the lesion (mm2) was calculated by outlining the missing cortical tissue for each section taken at 1 mm intervals (MCID, Imaging Research, Ontario, Canada), and the volume (mm3) of the lesion was determined by taking the sum of the contusion areas obtained from each section multiplied by the distance between sections (1 mm).
Western blotting: Sample preparation
Three weeks after CCI or sham injury, rats [TBI + CV (n = 6), TBI + CD (n = 6), TBI + AV (n = 6), TBI + AD (n = 6)] underwent rapid decapitation and gross dissection of the frontal cortex and HC. After tissue dissection, samples were snap-frozen in liquid nitrogen and stored at −80°C for later analysis. In preparation for Western blotting, frontal cortex and HC tissue was placed in 5–10 mL lysis buffer [0.1 m NaCl, 0.01 m Tris-Cl, 0.001 m EDTA, 1 μg/mL aprotinin, 100 μg/mL phenylmethylsulfonyl fluoride (PMSF)]. Samples were sonicated for 30s at 30mV. The lysates were centrifuged at 13,800g for 30 min at 4°C. The supernatant fluid was then divided into aliquots and frozen at −80°C.
Western blotting
The plasticity proteins analyzed were growth-associated protein 43 (GAP-43) and synaptophysin. The protein concentration was measured by using a bicinchoninic acid (BCA) assay kit (Pierce, Rockfold, IL). Aliquots of 50 μg of protein from each sample were mixed with 2x sample buffer and boiled for 5 min. The proteins were resolved on a 10% SDS-polyacrylamide gel and transferred onto Hybond-PVDF membranes (Amersham, Arlington Heights, IL). Membranes were blocked using Tris-buffered saline with Tween-20 (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, and 0.05% Tween-20) containing 5% nonfat milk for 1 h, then probed with anti-synaptophysin antibody (1:10,000 Santa Cruz) for 1 h or anti-GAP-43 (1:1000). After washing four times with Tris-buffered saline with Tween 20, the membranes were probed with anti-mouse horseradish peroxidase conjugated antibody (1:20,000) to allow detection of the appropriate bands using enhanced chemiluminescence (Amersham, Arlington Heights, IL). The membranes were then stripped and probed again with anti-beta-actin antibodies (1:10,000) to assess protein loading. The band densities were analyzed by a NIH Scion image (Scion, Frederick, MD).
Statistical analysis
Data are presented as mean +/− SEM (standard error of the mean). For behavioral tasks that involved multiple test sessions (motor testing and MWM), analysis of variance for repeated measures (RMANOVA) was utilized. Tests for sphericity were evaluated for within-subject comparisons and corrected for violations using the Greenhouse–Geisser test. Post hoc contrasts were set to make pair-wise comparisons between injury groups. To assess motor and MWM performance on a specific testing day, single factor analysis of variance (ANOVA) with pair-wise comparisons between injury groups were made as described above. Multivariate analysis of variance (MANOVA) was used to assess main effects observed for y-maze performance. Paired t-test was used for within-group comparisons of novel versus familiar arm exploration in the y-maze task. Histological comparisons were made using Student's t-test without equal variance assumed. Neuroplasticity markers were assessed using MANOVA. As there were no significant differences in protein loading, group differences in GAP-43 and synaptophysin levels were evaluated with band density reported as a percentage of sham. Statistical analyses were completed using SPSS for Windows, version 14.0.
Results
Motor testing
In the chronic treatment experiment, injured rats were transiently impaired on the beam balance task. There was a significant injury effect, day effect, and injury*day interaction (p < 0.003 all comparisons) for beam balance performance. There were no significant differences between Dilantin- and vehicle-treated injury groups. All injury groups returned to baseline by day 5 of testing (Fig. 1A). Beam walking latency and beam walking score showed an injury effect, day effect, and injury*day interaction (p < 0.0001 all comparisons). Overall there were no Dilantin-related effects on beam walking performance. However, on day 6, the TBI + CD group performed significantly worse than the TBI + CV group for both walking latency and score (p = 0.001 all comparisons), and they did not return to baseline by day 6 of testing (Fig. 1 B,C).

Motor Performance Tasks. Chronic Dilantin Administration: (
Results in the acute-treatment experiment were similar to those observed in the chronic-treatment experiment. For beam balance, there was a significant injury effect, day effect, and injury*day interaction (p < 0.003 all comparisons). No significant differences between Dilantin- and vehicle-treated injury groups were noted, and injury groups returned to baseline by day 5 of testing (Fig. 1D). Beam walking latency and score showed an injury effect, day effect, and injury*day interaction (p < 0.0001 all comparisons). Overall there were no Dilantin-related effects on beam walking performance. However, on days 3 and 4, the TBI + AD group performed worse, on both latencies and scores, than did the TBI + AV group (p < 0.04 all comparisons). Both the TBI + AV and TBI + AD groups returned to baseline by day 6 of testing (Fig. 1 E,F).
Y-maze
With the chronic treatment groups, for y-maze performance, there was a significant arm effect (p < 0.001) and injury*arm effect (p < 0.0001) was seen. No treatment*injury interactions were found. Increases in novel arm vs. familiar arm exploration were noted in sham groups only (p < 0.05 all comparisons, Fig 2A). Y-maze results in the acute-treatment experiment show a significant arm effect (p < 0.0001) and injury*arm interaction (p < 0.02). There were no treatment*injury interactions found. There was a trend for increased novel arm exploration in the TBI + AD group (p = 0.06), and both sham groups demonstrated a significant preference for the novel arm (p < 0.05). Paired t-tests show the injury*arm interaction is driven by a lack of statistical differences in familiar vs. novel arm exploration in the injury vehicle group (Fig. 2B).

Y-Maze Task.
MWM
There was a significant effect of group and days (p < 0.0001 all comparisons) on latency to find the hidden platform and path-length taken to find the hidden platform among the chronic- treatment groups. The TBI + CD group performed significantly worse overall than the TBI + CV group with regard to both latency and path-length taken to find the hidden platform (p < 0.03 both comparisons). Significant differences for latency and path-length taken were noted on post-surgery days 14 and 15 (p ≤ 0.05 all comparisons) between the TBI + CD group and the TBI + CV group
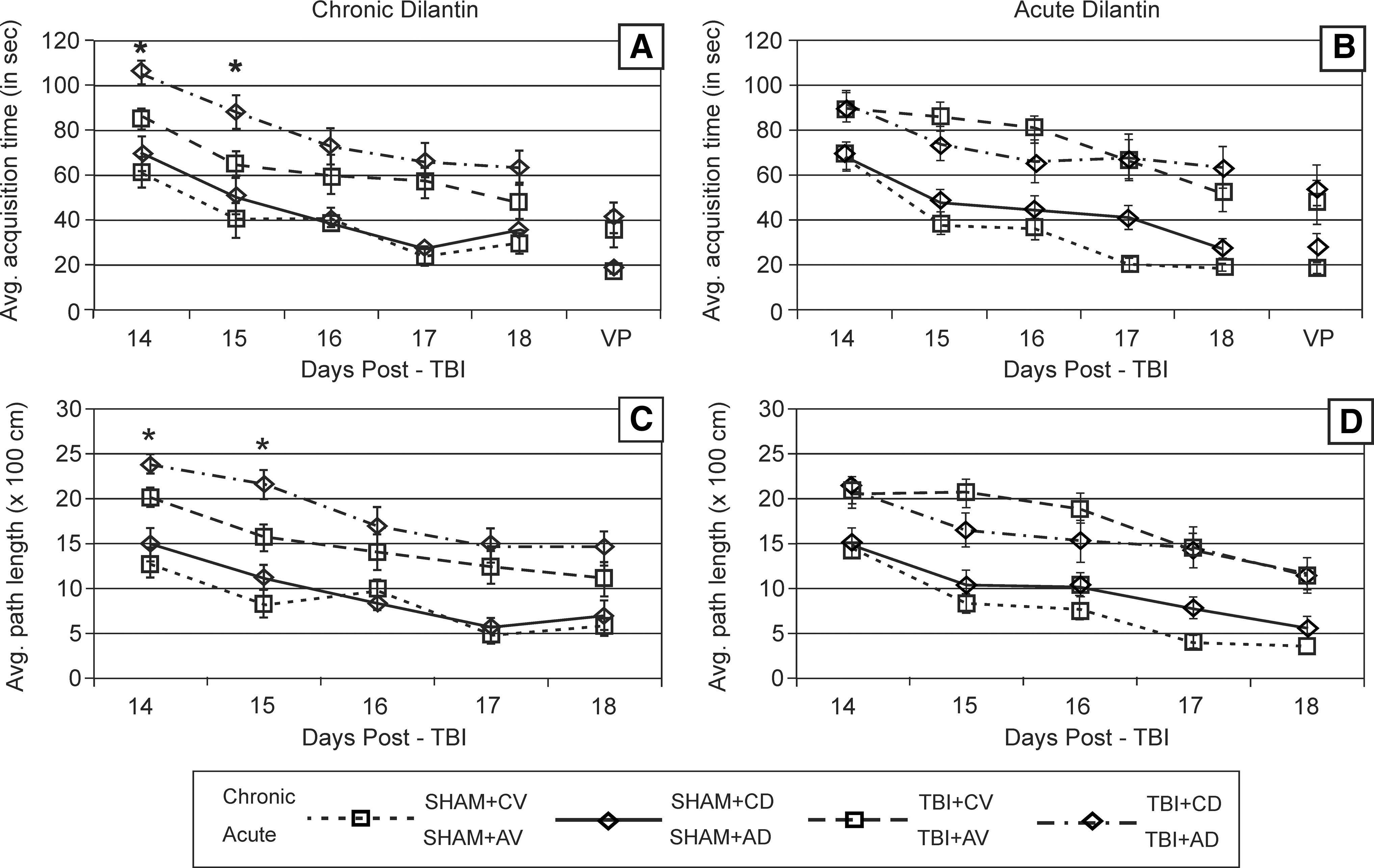
Morris Water Maze Task
HC cell survival
All injured rats in both the chronic- and acute-treatment experiments had a significant loss of morphologically intact neurons in the HC on the hemisphere ipsilateral to the injury site. The TBI + CD group had a significantly lower percent survival (ipsilateral/contralateral) of CA1 neurons than did the TBI + CV group (p < 0.035). There was no difference in CA3 neuron survival between injury groups (Fig. 4A). In contrast, the TBI + AD group had a significantly higher percent survival of CA1 neurons than did the TBI + AV group (p = 0.035). There was no significant difference in CA3 neuron survival (Fig. 4B).
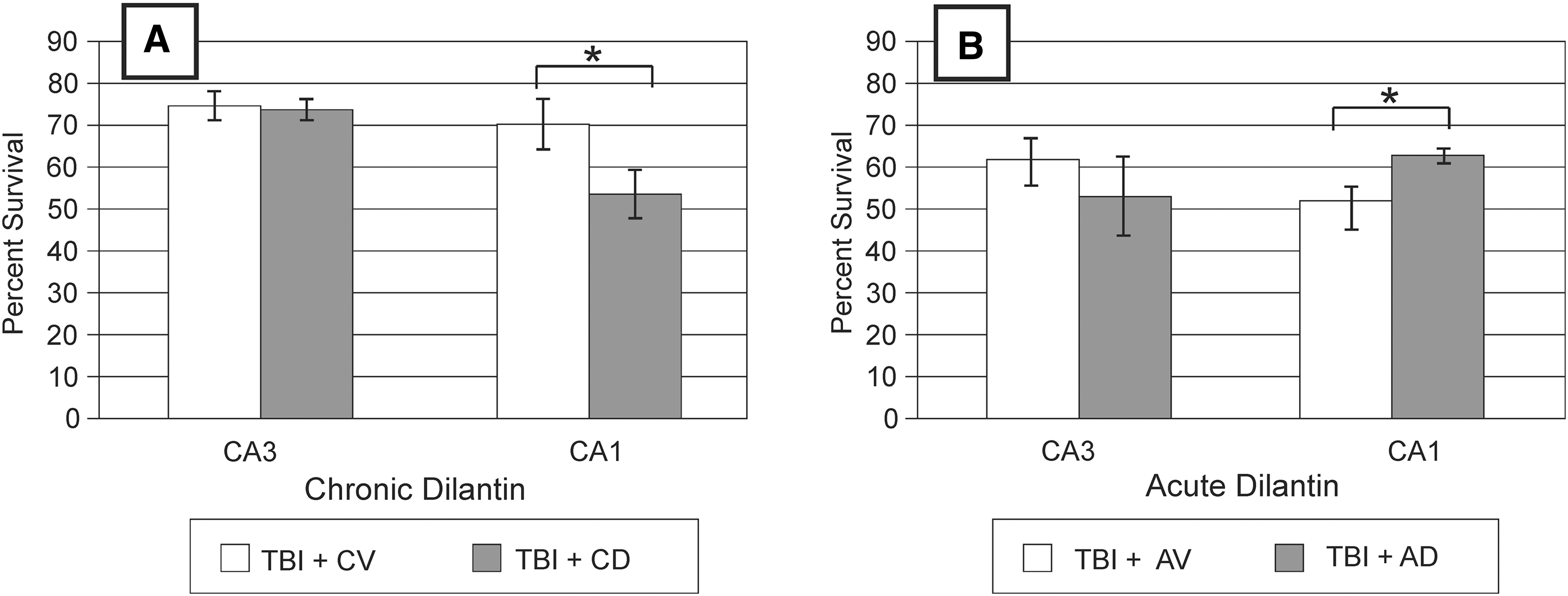
Hippocampal Neuron Survival
Cortical contusion volume
The mean cortical contusion volume of the TBI + CD group was 32.71 ± 2.31 mm3 and the mean cortical contusion volume of the TBI + CV group was 37.60 ± 1.82 mm3. Although the mean for the TBI + CV group was somewhat higher, it did not reach statistical significance (p = 0.060). The mean cortical contusion volume of the TBI + AD group was 42.06 ± 3.47mm3 and the mean cortical contusion volume of the TBI + AV group was 42.33 ± 3.42 mm3. The two were not statistically different (p = 0.478).
Western blotting
For rats receiving chronic daily Dilantin treatment, there was a significant injury effect (p < 0.0001) but no treatment effect for GAP-43 expression in the ipsilateral HC (Fig. 5A). There also was a significant injury effect on synaptophysin levels in the HC ipsilateral to the injury site (p < 0.0001), however, chronic Dilantin treatment did not affect synaptophysin levels in this region (Fig. 5C). There were no significant differences between groups in either protein in the contralateral HC (Fig. 5B,D

Neuroplasticity Markers, Chronic Dilantin Treatment.
For rats receiving acute Dilantin treatment, there was a significant injury effect (p < 0.0001) and a significant treatment effect (p = 0.008) for GAP-43 expression in the ipsilateral HC. Whereas injury reduced GAP-43 expression, acute treatment increased GAP-43 for both rats with CCI and those with sham surgery, as compared to the respective vehicle control groups (Fig. 6A). There also was a significant injury effect on synaptophysin levels in the HC ipsilateral to the injury site (p < 0.0001), however, acute Dilantin treatment did not affect synaptophysin levels in this region (Fig. 6C). There were no significant differences between groups in either protein in the contralateral HC

Neuroplasticity Markers, Acute Dilantin Treatment.
Serum Dilantin levels and weights
The serum drug levels taken from the SHAM + CD group were minimal (0.35 ± 0.17 μg/mL.). There were no detectable serum drug levels in the SHAM + CV group. Weight gain post-surgery was modestly reduced for the SHAM + CD and TBI + CD groups compared to and rats with TBI and sham surgery that were receiving vehicle. On average the SHAM + CV group took 3 days and the TBI + CV group took 5 days to return to 100% of pre-injury weight. The SHAM + CD group took 8 days and the TBI + CD group took 9 days to return to 100% of pre-injury weight. There were no significant differences among any injury or sham groups receiving acute vehicle or acute Dilantin (data not shown).
Discussion
Although Dilantin therapy is a commonly used treatment strategy for PTS prophylaxis with clearly established guidelines, there remains inconsistency in clinical practice. The AAN currently recommends a limited treatment course with Dilantin (i.v) in patients with moderate-to- severe brain injury (Chang and Lowenstein, 2003). Despite these recommendations, many TBI patients receive Dilantin (or other anticonvulsants prescribed for PTS prophylaxis) well beyond 7 days post-injury. We hypothesized that, during the acute phase after TBI, the mechanisms by which Dilantin confers its anticonvulsant properties would also be neuroprotective. We also hypothesized that the same mechanisms by which Dilantin might be neuroprotective in the acute phase would, when given chronically, be detrimental to recovery.
To our knowledge, the effects of Dilantin administration on neural recovery after CCI have not been previously explored. Our results from the beam walking tests suggest that the CCI group receiving acute Dilantin treatment had transiently increased impairment in the days following removal of the drug. This temporary increase in deficits is temporally consistent with the pharmacokinetic properties of Dilantin previously described (Rundfeldt and Loscher, 1993). In contrast, Dilantin does not appear to have adverse effects on the chronic daily treatment group until day 5, and is statistically significant by day 6, when CCI rats treated with Dilantin did not return to their baseline level of function with this task. Chronic Dilantin after TBI had no effect on exploratory behavior but worsened MWM performance and reduced CA1 neuron survival at 21 days post-CCI, when compared to vehicle administration. Conversely, acute Dilantin administration modestly improved exploratory behavior, had no adverse affect on MWM performance, improved CA1 neuron survival, and increased HC GAP-43 levels post-CCI compared to vehicle administration.
In addition to the initial insult, patients with TBI are also at risk for secondary injury and other complications including PTS. Previous reports demonstrate that Dilantin provides some degree of neuroprotection in other experimental models of brain injury such as ischemia (Boxer et al., 1990; Brown et al., 1995; Chan et al., 1998; Taft et al., 1989), and ischemia from concomitant injury and hemorrhagic shock can be an important component to overall pathology associated with TBI (Dennis et al., 2009). The excitatory neurotransmitter, glutamate, is found in high concentrations in the extracellular space and CSF soon after TBI and remains elevated for at least a week (Wagner et al., 2005; Zhang et al., 2001). Elevated extracellular glutamate levels after TBI are a primary contributor to excitotoxic injury. Choi has suggested two mechanisms of glutamate mediated cell death: 1) an influx of Na+ and Cl- causing cell vacuolization and swelling; and 2) the influx of Ca++ which leads to delayed damage (Choi, 1987 a,b). Calcium dysregulation has been proposed as the final common pathway of cellular destruction (Siesjo, 1993).
The pharmacological properties of Dilantin that explain its anticonvulsant effects also provide theoretical support for potential neuroprotection against sodium and calcium dysregulation associated with excitotoxicity if it is administered soon after TBI (Chweh et al. 1986; Cunningham et al., 2000; Ragsdale and Avoli, 1998; Schumacher et al., 1998; Sitges et al., 2007). However, excitatory neurotransmitter systems are depressed later after injury and are required for synaptic plasticity and recovery of function during the post-acute phase of TBI. In addition, clinical studies show that Dilantin is not beneficial in preventing late-onset seizures (Temkin et al., 1990). Moreover, TBI patients who are given ongoing Dilantin treatment have significantly worse neurobehavioral deficits 1 month post-TBI when compared to placebo treated patients in motor function/planning, attention, concentration, memory, verbal skills, reasoning, and psychosocial performance (Dikmen et al., 1991).
Dilantin has dose- and duration-dependent neurological adverse effects that include CNS depression, confusion, alteration of sleep/wake cycles, ataxia, and dyskinesias. In the clinical setting, Dilantin is often administered at multiple time points daily to maintain serum levels in the therapeutic range. However, metabolism of the drug in rats is much more complex and makes it challenging to maintain serum levels in the therapeutic range. Rundfeldt and Loscher have shown that daily injections of 75 mg/kg of Dilantin result in neurotoxic effects in 2–3 days (Rundfeldt and Loscher, 1993). The half-life of the drug was 4 h after the first injection, and about 20 h for the second and third injections. The first dose led to long-lasting saturation of Dilantin-metabolizing enzymes such that subsequent doses caused systemic accumulation of the drug. Further study showed that metabolizing enzymes remained inhibited even though the Dilantin was already eliminated. Based on their findings, a treatment protocol in that used 75 mg/kg i.p. on the first day, and 50 mg/kg daily on subsequent days was established as best achieving therapeutic levels (Rundfeldt and Loscher, 1993). We adopted this protocol and performed behavioral testing 24 h post- injection. Trunk blood 24 h after the last injection showed that serum Dilantin levels were minimal and ensured that behavioral testing for the chronic-treatment study was not confounded by supra-therapeutic or toxic serum Dilantin levels.
The beam balance and beam walking tasks administered to the CCI model capture transient deficits that typically return to baseline 5–6 days post-injury (Dixon et al., 1996, 1998, 1999b). Other studies have reported that factors such as gender (Wagner et al., 2004), environmental enrichment (Kline et al., 2007), and hypothermia treatment (Dixon et al., 1998; Kline et al., 2002) can impact performance on these motor tasks. In acute-dosing studies with cats, Dilantin has been shown to have a negative impact on performance on beam balancing and walking tasks (Carp and Anderson, 1979). Also, Dilantin administration in rats after a somatomotor cortex lesion is deleterious to motor performance on a narrow beam walking task (Brailowsky et al., 1986). These study findings are consistent with the early Dilantin effects associated with increased motor deficits in our CCI model. Beam walking performance, but not beam balance performance, was affected by Dilantin treatment. In contrast to multiple studies suggesting neuroprotection for subcortical structures (Chan et al., 1998; Reckling, 2003; Taft et al., 1989), it is not clear why Dilantin has negative effects on motor tasks generally, and in relation to cortical lesioning and now CCI, specifically. The beam walking task requires more skilled motor activity, and the TBI + CD group did not return to baseline by the last day of testing. However, other tasks used in this experiment (y-maze and MWM) do not require skilled motor control and therefore were not confounded by skilled motor impairments. Swimming speeds measured during the MWM revealed no statistical differences between injury groups, suggesting that motor abilities did not confound results for this task. Although swimming speeds do not reflect skilled motor deficits at 2 weeks post-injury, it would be interesting to further evaluate the time course of eventual return to baseline with the beam walking task and the chronic daily Dilantin dosing regimen.
It has been widely shown that experimental TBI also produces spatial learning acquisition deficits in the MWM (Bramlett et al., 1997; Dixon et al., 1999 a,b; Hoffman et al., 1994; Kline et al., 2000; Scheff et al., 1997). Additionally, MWM deficits are sensitive to treatment interventions following experimental TBI, a few of which include environmental enrichment (Kline et al., 2007; Wagner et al., 2002), methylphenidate treatment (Kline et al., 2000; Wagner et al., 2007b), and hypothermia (Bramlett et al., 1995; Dixon et al., 1998). Studies in uninjured rats have also shown that twice-daily chronic treatment with Dilantin produces deleterious performance impairments in the MWM (Churchill et al., 2003). We found that chronic, but not acute, Dilantin therapy results in a worsening of spatial learning after CCI. Rats receiving daily Dilantin showed significantly worse performance in the MWM on days 14 and 15 individually, as well as in overall analysis when compared to the vehicle group. Findings were significant for both latency and path-length analysis. However, time spent in the circle encompassing the escape platform during acquisition trials was only influenced by CCI status and not by Dilantin therapy during the probe trial. We completed behavioral testing each day prior to daily Dilantin administration. This point, along with the fact that swimming speeds were unaffected by treatment and that serum Dilantin levels were negligible when measured 24 h after the last daily dose given, provides adequate evidence for one to conclude that behavioral deficits associated with daily Dilantin therapy were the result of negative effects on neural recovery rather than of sedation that could be associated with elevated or toxic drug levels.
The y-maze task is a voluntary exploration task that allows the observer to examine an animal's exploratory behavior and novel arm preference after habituation in two arms of the maze (Dellu et al., 2000) and is sensitive to the effects of CCI, which causes novel arm exploration to be diminished (Wagner et al., 2007a). Although Dilantin has not been well studied with regard to its effects on exploratory behavior, we show that rats with CCI given acute Dilantin treatment experienced a modest benefit in novel arm exploration in the y-maze. This benefit may be attributable to the neuroprotective effect of acute treatment, and may represent relatively preserved spatial recognition memory, consistent with HC sparing associated with acute Dilantin treatment. Alternatively, benefits may be attributable to increased innate exploratory drive.
Acute Dilantin treatment can result in neuronal sparing in models of brain injury using ischemic stroke (Boxer et al., 1990; Brown et al., 1995), in vitro models of ischemia (Rekling, 2003) and spinal cord injury (Ates et al., 2007). In addition to our behavioral assessments, we evaluated the efficacy of Dilantin as a neuroprotective agent on HC cell survival and cortical contusion volume. Acute Dilantin dosing resulted in a significantly greater sparing of HC CA1 neurons ipsilateral to the injury site than did vehicle treatment. In contrast, the chronic Dilantin paradigm resulted in a significantly lower percent survival of HC CA1 neurons brain ipsilateral to the injury site than in the vehicle-treated group. Whereas these observations support the role of Dilantin as a neuroprotective agent during the acute post-injury period, chronic dosing failed to confer neuroprotection and also had deleterious effects on CA1 neuron survival following CCI. Our data do not, however, provide a mechanism for this observation or indicate the timing in which this transition from cell sparing to cell loss occurs. Additional time course studies would be helpful here, not only to allow us to understand the mechanism and timing of this phenomenon, but also to determine the functionality of spared HC neurons and any pro-convulsant properties.
To measure axonal reorganization and synaptic plasticity, we analyzed two pre-synaptic proteins, GAP-43 and synaptophysin. GAP-43 is a phosphoprotein associated with neuronal growth, formulation of novel neuronal connections, synaptic remodeling, and neuronal sprouting following traumatic insult (Benowitz and Routtenberg, 1997). Synaptophysin is linked to reactive synaptogenesis in sprouting neurons (Bergmann et al., 1997). With the chronic dosing paradigm we found that GAP-43 expression in the ipsilateral HC was significantly higher in the sham vehicle group than in both the TBI chronic vehicle and Dilantin groups. However, GAP-43 expression in the ipsilateral HC was somewhat lower in the TBI group receiving chronic Dilantin compared to the TBI group receiving chronic vehicle. In contrast, acute Dilantin treatment resulted in increased GAP-43 levels compared to acute vehicle treated sham controls. With both experiments, HC synaptophysin levels were significantly lower in the ipsilateral hemisphere for all of the injury groups than for the sham vehicle groups. Taken together, these results suggest that acute, but not chronic, Dilantin dosing improves injury-related decreases in HC markers of neuroplasticity. Importantly, increases in HC GAP-43 expression in rats receiving acute Dilantin dosing is consistent with HC cell sparing also noted with this treatment group.
Although the findings of this study provide multiple lines of evidence suggesting that prolonged daily Dilantin therapy should be avoided where possible after TBI, some limitations and need for further research exist. For these experiments, we carefully chose a Dilantin dosing regimen for rat CCI intended to emulate the 1 week clinical dosing regimen endorsed by the AAN. Although precise correlates between rodent models of TBI and human TBI regarding timing of secondary injury cascades are not entirely clear, and given the extended pharmacokinetic properties of Dilantin in rats after the first dose, we felt that our acute-dosing regimen in rats was a reasonable representation of a 1-week Dilantin treatment paradigm in the clinical human population. Similarly, the daily dosing regime was a reasonable representation of the common clinical presentation of prolonged Dilantin therapy for prophylaxis that extends into the post-acute inpatient rehabilitation period and beyond. However, more work should be done exploring a dose-response paradigm, and exploring if there are alternative chronic-dosing regimes that do not result in adverse effects on neurological and behavioral recovery after TBI. Previous clinical work suggests that some cognitive improvements occur after discontinuation of Dilantin (Dikmen et al., 1991). Further work should assess lasting influences of daily Dilantin on neurological and behavioral recovery after drug removal. Another potential limitation of this study was that it was not designed to monitor daily serum drug levels to ensure rats were maintained in the therapeutic range. However, the minimal levels noted 24 h after the last dose given 3 weeks after surgery supports the conclusion that there was not a significant accumulation of Dilantin over time, and that blood levels at the time of behavioral testing were not confounding behavioral performance. The overall effects of Dilantin on behavior, however, are likely to be minimized with this research design, which is important when one is thinking clinically about Dilantin effects in patients who are required to function with higher Dilantin levels in their system. Male rats were studied in this set of experiments. Given potential sex/hormone differences in neuroprotection and neuroplasticity after TBI (Roof and Hall, 2000; Stein, 2008), it is possible that there could be some sex/hormone-based differences in the neurobehavioral and neurobiological effects of both acute and chronic Dilantin therapy in female rats with CCI.
Conclusions
Prolonged daily Dilantin therapy has adverse effects on neurological indices of recovery and cognitive task performance in the CCI model of TBI. In contrast, a limited course of Dilantin therapy during the first 24 h after CCI does not adversely influence cognition and has neuroprotective effects on HC cell sparing and neuroplasticity markers. Based upon our findings, Dilantin administration for PTS prophylaxis in clinical populations should be limited to, and not extend beyond, the acute post-injury period. Additionally, future work should focus on identifying anticonvulsant medications that have a minimal adverse impact on recovery when clinical need requires post-acute dosing.
Footnotes
Acknowledgments
This work was supported by The Foundation for PM&R and the University of Pittsburgh Department of Physical Medicine and Rehabilitation.
Author Disclosure Statement
No competing financial interests exist.