
Abstract
Blast-induced traumatic brain injury (TBI) is the signature insult in combat casualty care. Survival with neurological damage from otherwise lethal blast exposures has become possible with body armor use. We characterized the neuropathologic alterations produced by a single blast exposure in rats using a helium-driven shock tube to generate a nominal exposure of 35 pounds per square inch (PSI) (positive phase duration ∼4 msec). Using an IACUC-approved protocol, isoflurane-anesthetized rats were placed in a steel wedge (to shield the body) 7 feet inside the end of the tube. The left side faced the blast wave (with head-only exposure); the wedge apex focused a Mach stem onto the rat's head. The insult produced ∼25% mortality (due to impact apnea). Surviving and sham rats were perfusion-fixed at 24 h, 72 h, or 2 weeks post-blast. Neuropathologic evaluations were performed utilizing hematoxylin and eosin, amino cupric silver, and a variety of immunohistochemical stains for amyloid precursor protein (APP), glial fibrillary acidic protein (GFAP), ionized calcium-binding adapter molecule 1 (Iba1), ED1, and rat IgG. Multifocal axonal degeneration, as evidenced by staining with amino cupric silver, was present in all blast-exposed rats at all time points. Deep cerebellar and brainstem white matter tracts were most heavily stained with amino cupric silver, with the morphologic staining patterns suggesting a process of diffuse axonal injury. Silver-stained sections revealed mild multifocal neuronal death at 24 h and 72 h. GFAP, ED1, and Iba1 staining were not prominently increased, although small numbers of reactive microglia were seen within areas of neuronal death. Increased blood–brain barrier permeability (as measured by IgG staining) was seen at 24 h and primarily affected the contralateral cortex. Axonal injury was the most prominent feature during the initial 2 weeks following blast exposure, although degeneration of other neuronal processes was also present. Strikingly, silver staining revealed otherwise undetected abnormalities, and therefore represents a recommended outcome measure in future studies of blast TBI.
Introduction
B
Ling and associates (2009) recently reported that victims of severe blast TBI often exhibit complex brain injuries as revealed by cranial computed tomography, including acute refractory brain swelling and vasospasm. Armonda and colleagues (2006) also reported vasospasm in 47% of TBI victims in OIF, with enduring vasospasm seen in many patients. However, despite these seminal reports, the neuropathology of human blast TBI remains unclear across the injury spectrum.
Animal models of blast TBI have provided some insight into associated neuropathologic alterations. At the ultrastructural level, Kaur and co-workers (1995) found “darkened dendrites” associated with neurons in the cerebral and cerebellar cortices in the initial 14 days after explosive blast injury (110 kg TNT equivalent) in rats positioned in a concrete bunker simulating combat exposure. In this model, which produced ∼10% acute mortality, microglial activation was the most prominent feature. Cernak and colleagues (2001) reported electron microscopic evidence of vacuole formation in both nerve terminals in the hippocampus and myelin in rat brains studied at 24 h or 5 days after either whole-body blast exposure (a shock-tube model using ∼49 PSI) or isolated chest exposure (∼64 PSI). Increased pinocytosis and cytoplasmic extrusion of endothelial cells was also seen. These data suggested that an important component of brain injury resulted from extracerebral exposure. Saljö and associates (2000) exposed unprotected rats to blast TBI using explosive blast in a shock tube (a 2-g charge producing ∼35 PSI). Using an immunohistochemical approach with an antibody identifying phosphorylated neurofilament protein, immunoreactivity was detected in the temporal, cingulate, and piriform cortices, the dentate gyrus, and CA1 at 48 h and 7 days. They suggested that these findings reflected disturbed anterograde axonal transport with accumulation of phosphorylated neurofilament protein in perikarya. Moochhala and associates (2004) reported cortical neurodegeneration within Nissl-stained sections after a whole-body explosive blast exposure (2-g charge) in rats. In that study, treatment with aminoguanidine attenuated neurodegeneration, suggesting a detrimental role for nitric oxide synthase (NOS). More recently, Long and associates (2009) examined the effect of Kevlar body armor in a rat model of blast injury (shock tube, 18 or 21 PSI exposure), and reported that mortality was greatly reduced by body armor. In that study, the higher injury level also produced substantive evidence of global fiber tract degeneration as revealed by silver staining. At the 21-PSI injury level, fiber tract degeneration was accompanied by cortical necrosis ipsilateral to the exposure in some rats. Thus, Long and associates (2009) suggested that body armor could prevent otherwise lethal blast TBI in rats, and fiber tract degeneration was a key finding. However, some have posited that the effects of body armor on brain injury depend on the type of body protection used, and although reducing mortality, brain injury may be increased in some cases (reviewed by Cernak and Noble-Haeusslein, 2010). Svetlov and colleagues (2010) recently reported silver staining in deep brain regions after blast exposure to the head of rats (∼52 PSI) in a model with substantial head acceleration in studies that focused on serum biomarker assessments. In contrast, Risling and co-workers (2009) reported no evidence of axonal injury, as assessed with amyloid precursor protein (APP) immunohistochemistry, in rats exposed to explosive blast (20–34 PSI), despite observed deficits on beam walking and an altered gene array. Finally, Bauman and associates (2009) recently carried out extensive studies of explosive blast in a pig model. In moderate and severe blast exposures, the predominant pathological feature at 2 weeks after injury was fiber tract degeneration in the white matter of the cerebellum as revealed by silver staining. Glial fibrillary acidic protein (GFAP) staining was also prominent versus sham animals, suggesting secondary astrocytosis.
As part of the Defense Advanced Research Projects Agency (DARPA)-supported Preventing Violent Explosive Neurologic Trauma (PREVENT) blast program, we sought to characterize the neuropathological findings in a model of blast exposure injury (via shock tube) in rats with body shielding. These studies represent the initial characterization of the model to set the stage for a more comprehensive assessment of cellular, molecular, and biochemical mechanisms of injury—along with functional and additional neuropathologic characterizations. Our goal was to produce a moderately severe blast injury in rats with body shielding that was associated with survival in the majority of animals. We sought to determine if the neuropathologic alterations produced by this model would produce similar findings to those reported in the swine model of explosive blast TBI (Bauman et al., 2009).
Methods
Blast exposure
Twenty-six male Sprague-Dawley rats (350–425 g) from Charles River Laboratories were used in the study: 17 rats receiving blast exposures plus 9 sham animals. Using an institutional animal care and use committee (IACUC)-approved protocol, the bodies of isoflurane-anesthetized rats (2% isoflurane in air) were shielded in a steel wedge that was supported by a rod positioned 7 feet (2130 mm) from the end of a 21-foot (4270-mm) helium-driven shock tube (Fig. 1). A fully-formed decay profile was found to develop in the waveform at the test station (due to the reflected rarefaction from the driver), and there were no artifacts from either open-end rarefaction or contact surface in the blast simulation. Surface-flush PCB gauges were used for the shock measurements, because it is known that a gauge protruding or recessed by less than 1 mm will affect measurements. A diaphragm of 3×0.004” Mylar layers was used because it produced a faster and more complete rupture than a single 0.012” Mylar sheet; a sharp shock front had fully formed by the test station. The driver gas was helium charged into an initial volume of ambient air; this helium/air mix ensures a negligible density discontinuity at the contact surface interface between the expanding driver gases and shocked air, although in fact that interface did not affect the test station. Due to the inherent inefficiencies of plastic diaphragm rupture, the driver was pressurized to well above the value expected from theory. This shock tube generated a blast wave with a peak pressure of ∼35 PSI and a positive phase-duration of ∼4 msec. Figure 2 shows a shock-wave tracing that demonstrates the performance of the Mach wedge—confirming that a well-defined, uniform, and highly reproducible shock insult was imparted. The blast wave generated by a shock tube of this type is similar to the wave generated by high explosives. A steel wedge in the shock tube (Mach stem device in Fig. 1) was designed to protect the thorax and abdomen of the rat, as well as to enhance the shock-wave intensity. The design ensured that the triple point of the resultant Mach stem passed well above the head and thereby imparted a uniform shock-wave exposure. The apex of the wedge pointed toward the incident shock front, and at the apex, the incident shock front skimmed off the surface of the wedge, producing an amplified reflected wave—a Mach stem—that impinged on the left side of the rat's head. The ramp of this steel wedge was 8 inches (20 mm) wide at the base and 30 inches (76 mm) long, resulting in a ramp angle of 15 degrees. The rat's head was located 29.5 inches (75 mm) along the ramp face from the leading edge, with the head being 7 feet (2130 mm) from the shock tube muzzle and 14 feet (4270 mm) from the Mylar diaphragm.


Pressure (pounds per square inch, PSI) and time (msec) history generated during the initial 9 msec after blast exposure from a rat as produced using our shock-tube model. Surface flush PCB gauges were used for the recordings. This shock-wave tracing demonstrates the performance of the Mach wedge—confirming that a well-defined and uniform shock insult was imparted. For this example and all of the work presented in this report, we generated a 35 PSI exposure.
In order to assure that any ensuing brain damage would have been the result of the shock wave and trailing overpressure rather than due to rotational or impact forces, the rats' heads were backed against a compliant leather sling fitted between two short posts. This restraint method is comparable to the use of nylon mesh, and similar to methods employed by blast TBI experimenters to restrain gross motion of a specimen, yet preclude fixture-induced stresses. This approach was considered superior to restraining the head with hard backing, which potentially could have introduced blunt impact injury.
The blast exposure in the shock tube produced an injury with ∼25% mortality from apnea. Surviving rats were perfusion-fixed (hydrostatic pressure achieved via gravity) with phosphate-buffered paraformaldehyde (FD Neurotechnologies, Ellicott City, MD) at 24 h (6 rats), 72 h (5 rats), or 2 weeks (6 rats), with 3 sham rats being similarly perfusion-fixed at each of these three time points (9 shams total). After perfusion fixation, the dorsal calvaria were removed and the heads placed back into fixative. The heads were subsequently shipped in fixative solution to NeuroScience Associates (Knoxville, TN).
Histologic procedures
At NeuroScience Associates, the fixed brains were removed from the heads and treated with 20% glycerol and 2% dimethylsulfoxide to prevent freeze artifact. The brains were then multiply-embedded into two gelatin matrix blocks using MultiBrain Technology™. Fourteen brains were embedded in one of the two blocks and 12 within the second block. The brain blocks were allowed to cure and were then rapidly frozen by immersion in isopentane chilled to −70°C with crushed dry ice. Each of the two blocks were mounted on a freezing stage of a sliding microtome and sectioned coronally at a thickness of 40 μm. All sections were collected sequentially into an array of 4×6” containers that were filled with antigen preserve (buffered 50% ethylene glycol). At the completion of sectioning, each container held a serial set of one-of-every-24th section (i.e., one section every 960 μm). Sections from all 24 levels of both blocks (i.e., the brains from all 25 rats in the study) were stained with hematoxylin and eosin (H&E) and amino cupric silver, and with immunostains for glial fibrillary acidic protein (GFAP), ionized calcium-binding adapter molecule 1 (Iba1), and CD68 (ED1). Similar sections (from the same 24 brain levels) from one of the two brain blocks (representing 14 brains) were also immunostained for amyloid precursor protein (APP), and with an antibody against rat IgG, the latter to assess blood–brain barrier (BBB) permeability.
Amino cupric silver stain
The amino cupric silver staining closely followed the protocol described by deOlmos and associates (1994), with only an abridged version being presented here. The only step that was altered from deOlmos's procedure was the final bleaching step that uses Kodak rapid fix (specifically, elimination of the part B component, which is also the only step that is not described by deOlmos). Additional discussion of this stain is elaborated in an article that is available for download at the NeuroScience Associates website (Switzer, 2000). In brief, the 40-μm free-floating sections were taken through the following major steps: pre-impregnation, impregnation, reduction, bleaching, and fixing. The pre-impregnation solution contained cupric nitrate, silver nitrate, cadmium nitrate, lanthanum nitrate, neutral red, alpha-amino butyric acid, alanine, pyridine, triethanolamine, isopropanol, and deionized water (dH2O). After these components were well mixed, the solution was microwaved until it reached 45–50°C. The solution was then left to cool to room temperature and subsequently filtered. The sections were removed from the cacodylate-buffered formaldehyde and rinsed with dH2O. They were then placed in dishes containing the pre-impregnation solution and heated in the microwave to 45–50°C. The sections were then allowed to remain in this solution overnight to allow for cooling. The impregnation solution contained silver nitrate, 100% ethanol, acetone, lithium hydroxide, ammonium hydroxide, and dH2O. The sections were incubated in this solution for 50 min. The reducer solution contained 100% ethanol, formalin, citric acid, and dH2O. The sections were transferred from the impregnation solution into the reducer solution, and placed in a water bath with a temperature maintained between 32 and 35°C. After 25 min in the reducer solution, the sections were transferred into dH2O rinses. The bleaching solutions were potassium ferricyanide in potassium chlorate with lactic acid, potassium permanganate with sulfuric acid, and sodium thiosulfate. The sections were rapidly transferred through these bleaching solutions and then fixed in rapid fixer solution for 1.5 min. The sections were finally rinsed in dH2O, mounted on gelatin-subbed glass slides, and counterstained with neutral red to reveal normal cell bodies.
Immunostains
For all immunohistochemistry, free-floating sections were stained. All incubation solutions from the blocking serum onward used Tris-buffered saline (TBS) with 0.3% Triton-X100 (TX) as the vehicle; all rinses were with TBS. After a hydrogen peroxide treatment and blocking serum, the sections were incubated overnight at room temperature with the primary antibody. Following rinses, a secondary biotinylated antibody (anti-IgG of the host animal in which the primary antibody was produced) was applied. To visualize the location of the binding site of the primary antibody, an avidin-biotin-HRP complex was applied (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA). After rinses, the sections were treated with diaminobenzidine tetrahydrochloride (DAB) and hydrogen peroxide to create a visible reaction product. The sections were mounted on gelatinized (subbed) glass slides, air dried, dehydrated in alcohols, cleared in xylene, and cover-slipped. Negative controls were prepared and evaluated for each stain. The sources of the primary and secondary antibodies that were used are listed in Table 1.
Iba-1, ionized calcium binding adapter molecule 1 (a stain for microglia); GFAP, glial fibrillary acidic protein (a stain for astrocytes); CD68 (ED-1), a transmembrane protein expressed by tissue macrophages; APP, amyloid precursor protein (a membrane protein that concentrates in neuronal synapses and also represents an early marker of axonal injury); BBB, a stain for rat IgG used as a marker for a compromised blood–brain barrier; DAB, diaminobenzidine tetrahydrochloride.
Wako Chemicals USA, Inc., Richmond, VA; Dako North America, Inc., Carpenteria, CA; Serotec, Raleigh, NC; Millipore, Billerica, MA.
Illustrations
Photomicrographs were prepared using Nikon D300 and D700 cameras mounted on Olympus BH-2 microscopes. Photographs of full-coronal brain sections were taken using a Nikon D300 camera coupled with an Olympus 80-mm macro lens mounted on an Olympus Auto Bellows. At the time of image capture, slight adjustments were made to levels and sharpening with Nikon Capture NX2. Subsequent minor changes in Adobe Photoshop CS3 or CS5 included occasional additional image sharpening and the insertion of arrows, text, or magnification bars. No other image manipulations were performed.
Results
The intent of this article is to describe the general patterns of histologic alteration present within the brains of rats exposed to one form of blast injury, and to clarify which stains were found to be most helpful in delineating these patterns of injury. Because of the limited numbers of rats in each group and the large numbers of neuroanatomic regions examined, only qualitative severity scores are provided. Our purpose is to define the neuroanatomical alterations seen after blast exposure, and based on these targets, present a model that may be appropriate for future studies assessing cellular and molecular mechanisms and mitigation and/or treatment of blast injury.
Of the various stains employed in this study, the amino cupric silver stain was most sensitive for delineating all patterns of injury. Alterations in brain sections stained with amino cupric silver were primarily of three types: neuron degeneration, granular staining within the molecular layer of the dentate gyrus of the hippocampus, and axonal injury, with the last lesion representing the most consistent and persistent alteration found across the three sampling time points. (For one rat in the 2-week group, the blast tube malfunctioned, and it was suspected that this rat had received less than the full blast exposure. This rat was prospectively excluded from the study. Nevertheless, the brain from this rat was examined microscopically, and it did have evidence of a minimal degree of axonal injury within the cerebellar white matter.) The H&E stain and ancillary immunostains assisted in corroborating the presence of some of the histopathologic processes (especially degenerating neurons), but failed to reveal the potential for axonal damage to represent the primary pathologic process in this model of blast exposure in rats.
Neuron and/or dendritic degeneration
In sections stained with the amino cupric silver technique, degenerating neurons are stained black (i.e., are silver-positive), and are also characterized by staining and fragmentation of their cell processes, both axons and dendrites (Fig. 3). In this study, scattered foci of neuron degeneration were present at all three post-blast time points, but were less frequently found in rats necropsied at 2 weeks post-blast. Minimal to mild degrees of neuronal degeneration were present in 5 of 6 rats at 24 h, 4 of 5 rats at 72 h, and 2 of 5 rats at 2 weeks. Degenerating neuronal cell bodies did not have a consistent pattern of distribution within the brains but were found, for at least some rats, in the following neuroanatomic locations: entorhinal cortex (4 rats), frontal cortex (3 rats), CA1 pyramidal layer of the hippocampus (2 rats), cerebellar cortex (2 rats), parietal, piriform, insular, and temporal cortex (2 rats), and retrosplenial cortex (1 rat). Although definitively stained (i.e., degenerating), Purkinje neuron cell bodies were found in only 2 rats (Fig. 3C), and Purkinje neuron dendritic degeneration was quite common (Fig. 6A and B). In some but not all foci of neuron degeneration, small numbers of ED1 (CD68) cells were present, and Iba1 stains in these same regions showed slight evidence of microglial activation, although the magnitude of this response was only minimally above the threshold of sham animals (images not shown).

Examples of neuron degeneration seen in rats necropsied 24 h and 72 h following blast injury. All panels are from sections stained with amino cupric silver, so neurons and neuronal processes stained black are degenerative. In panel
Granular staining within the molecular layer of the dentate gyrus
Within amino cupric silver–stained sections of the hippocampus from blast-exposed rats, a fairly consistent pattern of granular staining was present within the middle portion of the molecular layer of the dentate gyrus of 3 of 6 rats necropsied at 24 h, and in 4 of 5 rats necropsied at 72 h post-injury (Fig. 4). However, this pattern of staining was not present in any of the rats necropsied at 2 weeks. This staining ranged from minimal to moderate in degree, with slightly higher severity scores (for degrees of staining) being assigned to rats necropsied at 72 h. For all affected rats, the staining was more prominent on the right side of the brain (contralateral to the blast exposure).
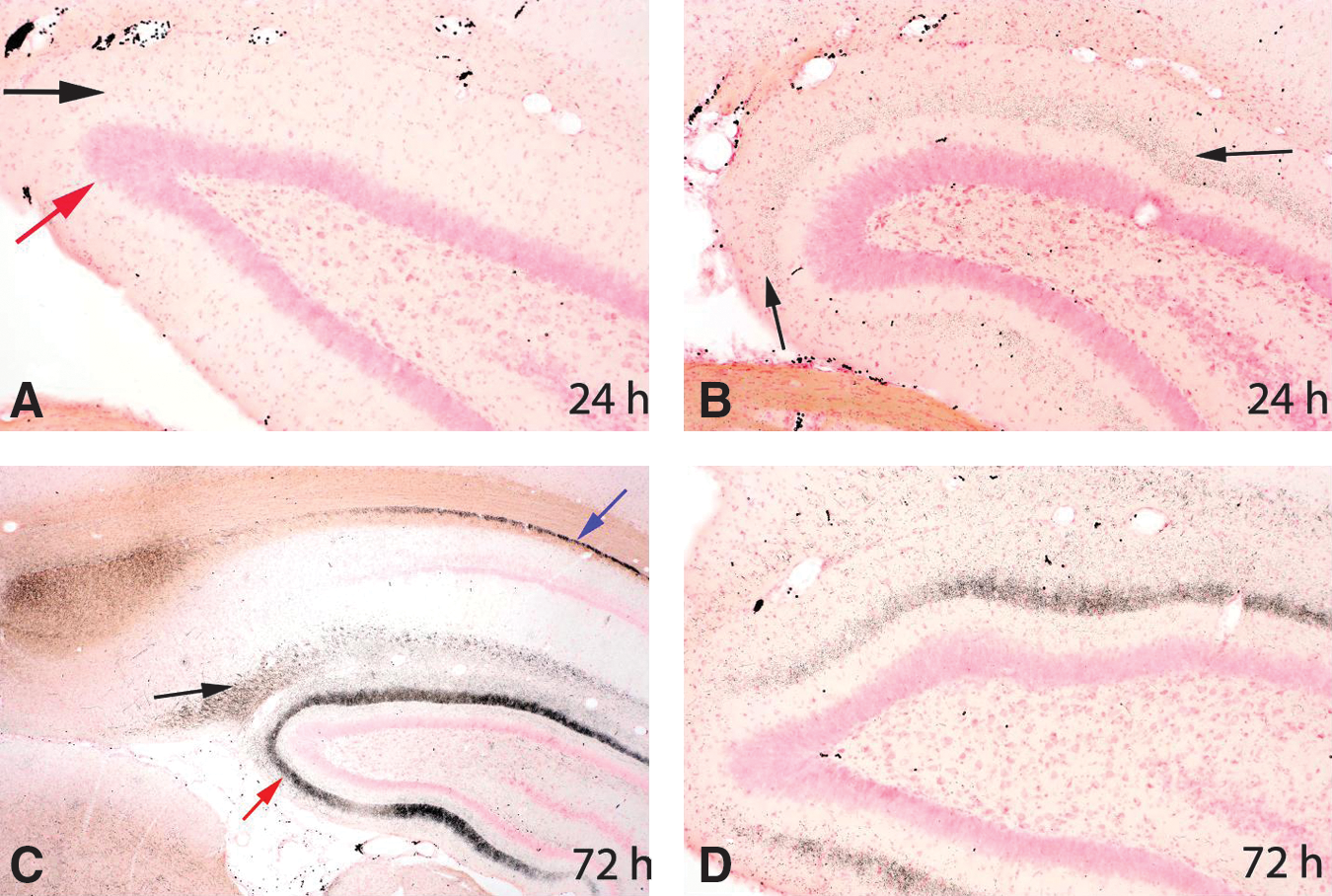
All sections are stained with amino cupric silver. All panels are of the dorsal hippocampal dentate gyrus from rats necropsied 24 h or 72 h post-injury. In panel
Axonal injury
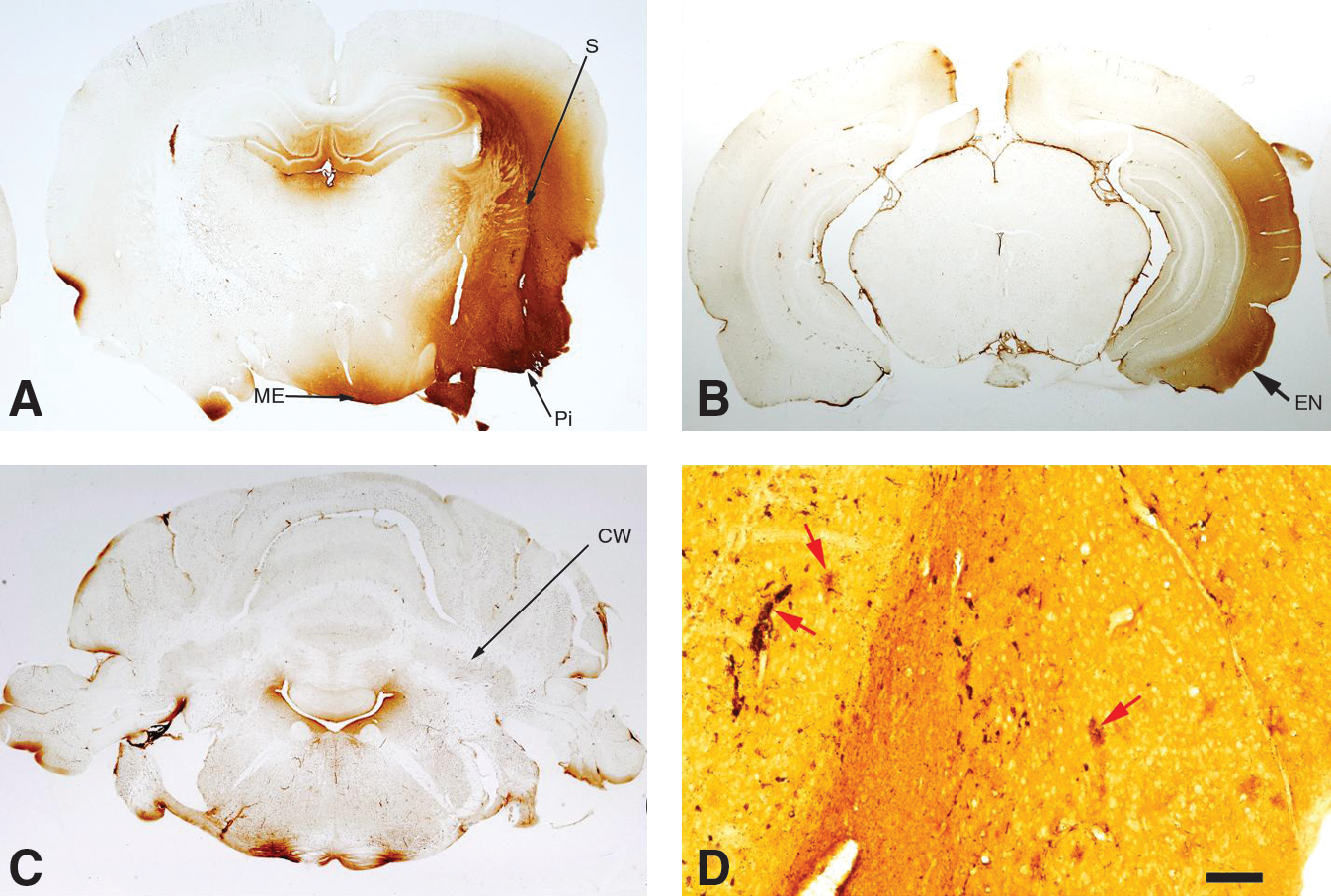
All blast-injured rats at all three time points (24 h, 72 h, and 2 weeks) had prominent axonal staining by amino cupric silver within the deep cerebellar white matter and brainstem regions (Figs. 5 –8). Axonal staining in the deep cerebellar white matter was sometimes symmetrical, but for numerous rats was more severe on the contralateral side of the brain (Figs. 5 and 6E). At 72 h and 2 weeks post-injury, scattered axonal spheroids in the affected regions were consistent with a diagnosis of diffuse axonal injury (Fig. 7D, E, and F). Although axonal injury was most consistently present within the cerebellar white matter, numerous other fiber pathways also showed damage (e.g., the optic nerve and tract, the internal and external capsules, the thalamic pathways, the cerebral and cerebellar peduncles, and the trigeminal tracts, trapezoid body, and pyramids). The optic nerve and optic tract were stained in all 5 of the 2-week rats (Fig. 7F), but in only one rat at 72 h, and in no rats at 24 h. Other white matter tracts showing degeneration extended from the level of the cortex to the medulla oblongata. Whereas evidence of neuronal degeneration was somewhat attenuated in the 2-week rats compared to those necropsied at 24 h and 72 h post-blast, axonal injury was most marked in rats from the 2-week necropsy.

Composite of five amino cupric silver–stained coronal sections through the middle of the cerebellum from each of the control, 24-h, 72-h, and 2-week post-injury groups. Black foci represent regions of axonal injury. Because of the very low magnification of these images, definitive axonal staining can only be observed in the most affected rats, but was present to some degree in the cerebellar white matter of all rats subjected to blast. Note that while injury appears to be bilaterally symmetrical for some rats (e.g., panels
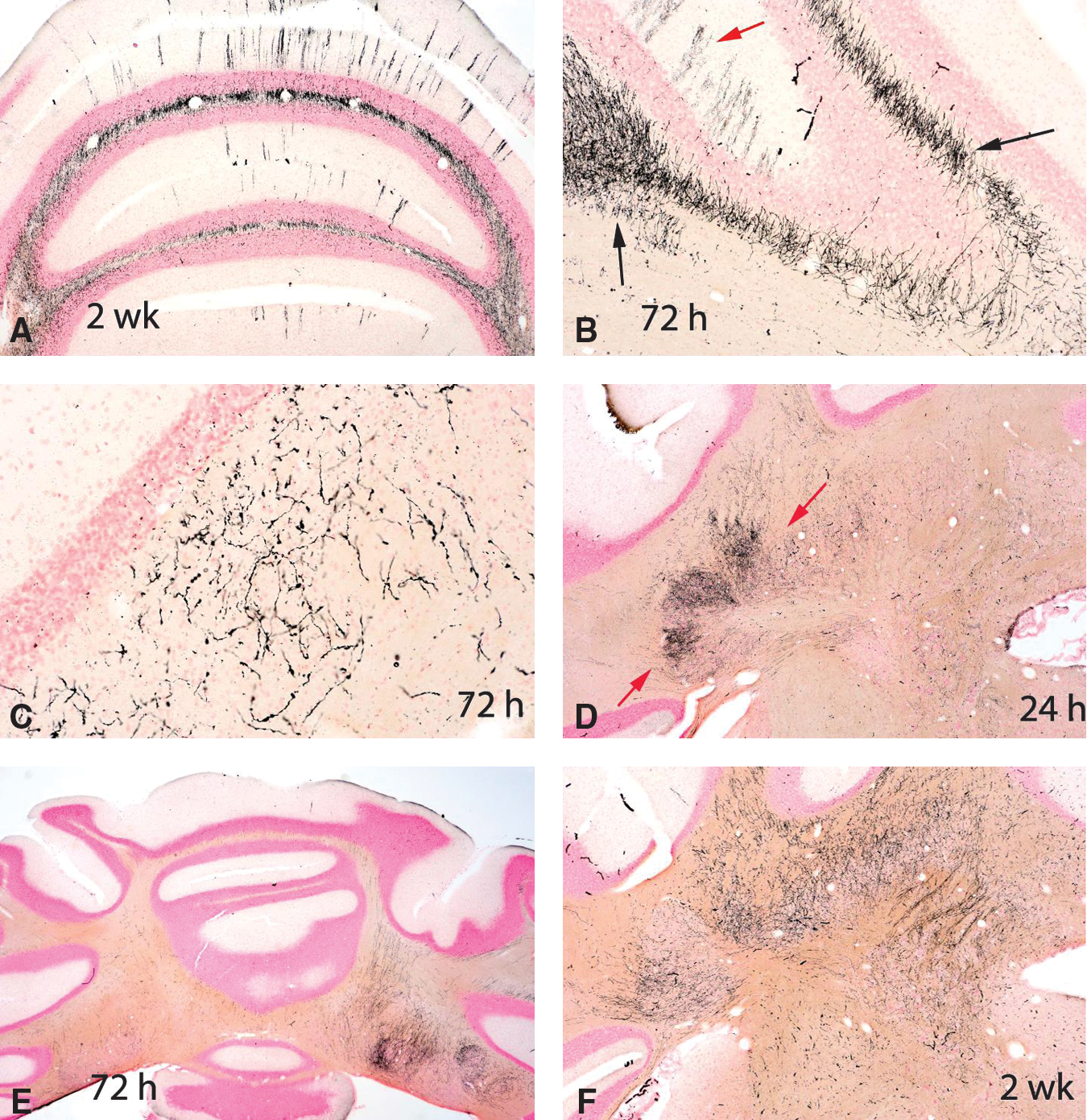
All six panels are of sections of cerebellum stained with amino cupric silver. Panel
All panels represent amino cupric silver–stained sections. These images indicate that axonal injury was present in a number of neuroanatomic regions (and not just in the cerebellum). Other affected regions included (but were not restricted to) the inferior colliculus (between the arrows in panel
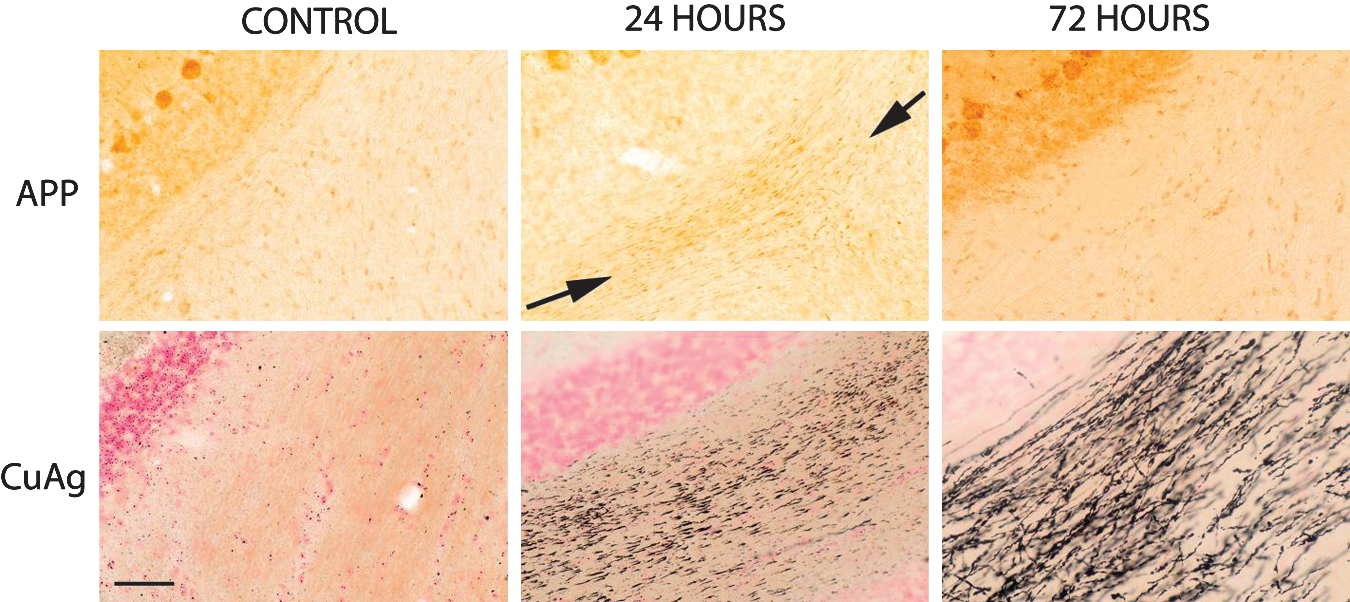
Contrast between the amyloid precursor protein (APP) and amino cupric silver (CuAg) stains in control rats and in rats necropsied 24 h and 72 h post-blast. All images were taken at the same magnification (20× objective), and each set of upper and lower APP and CuAg images was prepared from the same region of the cerebellum of the same rat. In contrast to the presence of extensive axonal staining by amino cupric silver at 24 h and 72 h post-blast, there is only slight axonal staining for APP at 24 h (between arrows), and little to no APP axonal staining at 72 h post-blast (scale bar=100 μm).
Immunostains
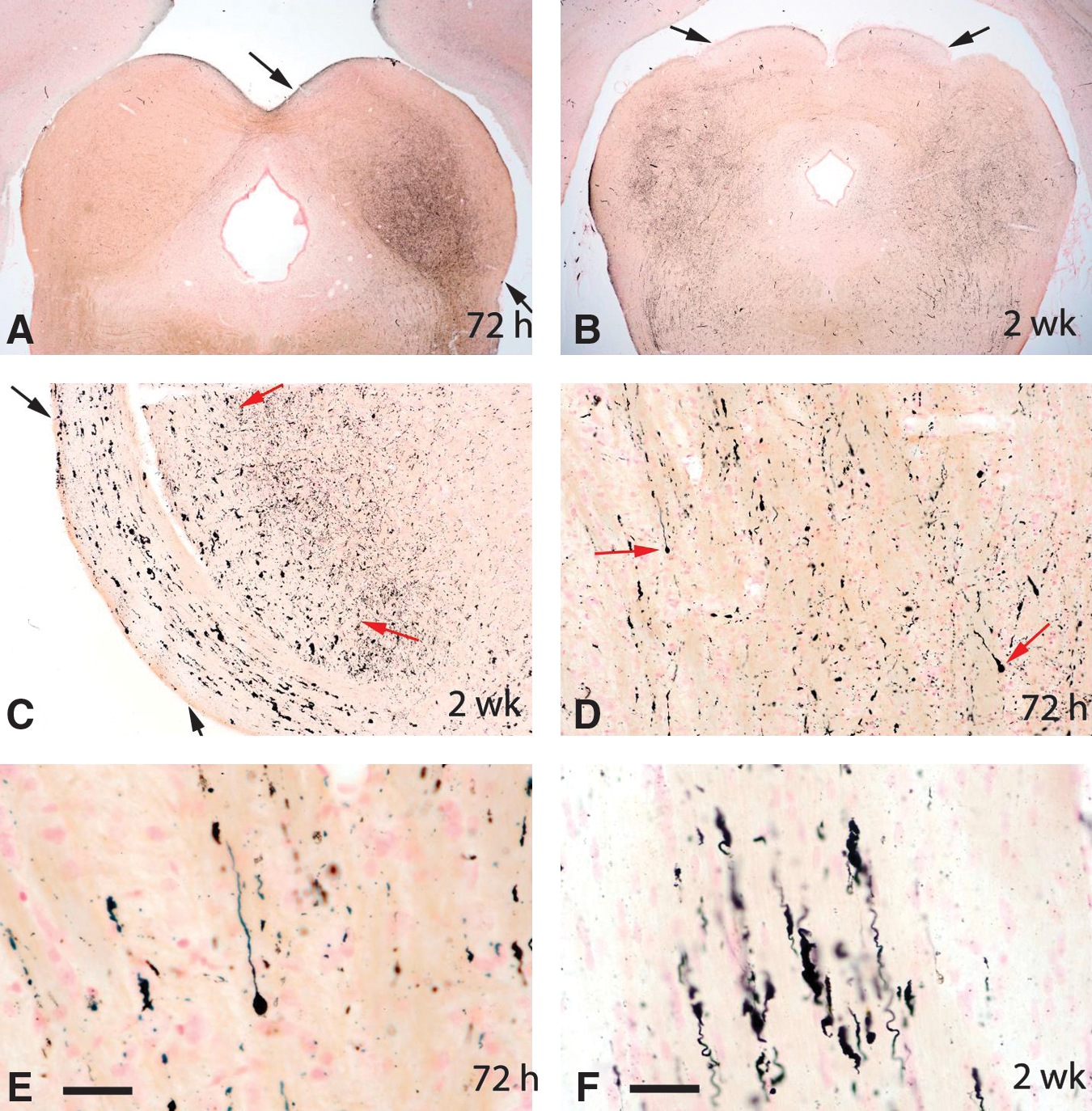
In spite of extensive evidence of axonal injury in amino cupric silver–stained sections, APP-stained sections typically showed only minimal axonal staining, except for the 24-h rats, which had mild axonal staining that was evident within the deep cerebellar white matter, and adjacent to some foci of acute neuronal degeneration (Fig. 8). Furthermore, GFAP staining failed to indicate any evidence of reactive astrocytosis in the brain regions characterized by extensive axon staining with amino cupric silver (Fig. 9). Iba1 staining was either not increased or only minimally increased in areas of injury demonstrated by amino cupric silver, although increased CD68 (ED1) staining was present in some foci of neuronal degeneration, and the Iba1 stain occasionally revealed a change in the morphology of small numbers of microglia, suggesting macrophage differentiation. BBB permeability as assessed by immunohistochemical staining for rat IgG demonstrated mild to moderate BBB compromise in the three 24 h post-blast rats on which this stain was performed. For these three 24-h rats, evidence of BBB leakage was primarily restricted to the contralateral side, and was principally limited to the neuropil of the piriform, entorhinal, temporal, and occipital cortices, as well as the underlying striatum (Fig. 10). However, no evidence of BBB leakage was present within the white matter regions where maximal axonal injury had been detected with the amino cupric silver stain (Fig. 10C). Furthermore, there was no evidence of BBB compromise in rats necropsied at 72 h or 2 weeks post-blast.
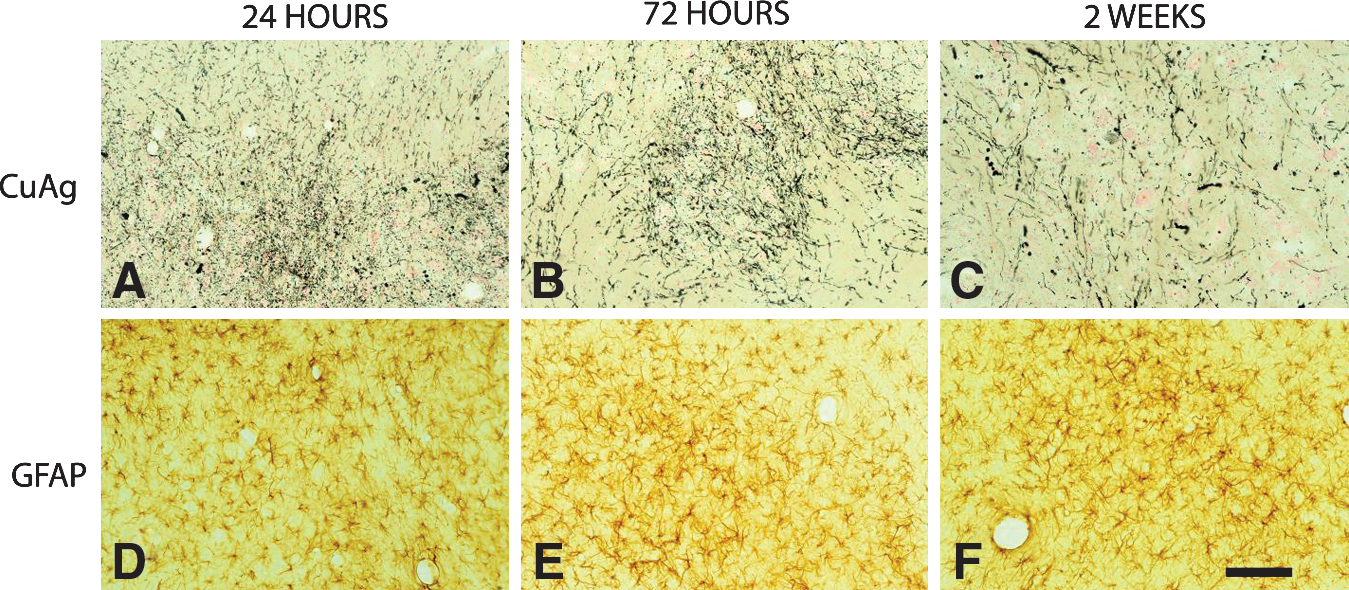
Axonal injury in the deep cerebellar white matter from rats necropsied at 24 h, 72 h, and 2 weeks post-blast. The top row of figures
All panels represent immunostains for rat IgG as an indicator of blood–brain barrier (BBB) compromise. Panels
Discussion
Our primary goal was to study neuropathologic alterations in the brains of blast-exposed rats that had body shielding. Our findings indicate that axonal, dendritic, neuronal, and synaptic (or axon terminal) degeneration, as demonstrated with the amino cupric silver stain, represent the most prominent neuropathologic features seen in the initial 2 weeks after blast injury in this shock-tube model of blast injury in rats with body shielding. Although some evidence of multifocal neuron degeneration was present within the brains of rats necropsied during the acute post-injury period (24 h and 72 h), by far the most consistent pattern of degeneration in this model involved the axons in the cerebellar white matter and in a variety of white matter tracts within the brainstem. Rather than becoming attenuated over time (as was the case for neuronal and synaptic/terminal degeneration), staining of axons with amino cupric silver was most prominent in the rats necropsied 2 weeks post-blast. Over time, there was also evidence of progression of the axonal degenerative process characterized by increased axonal fragmentation and axonal spheroid formation. These features are consistent with the process of diffuse axonal injury (DAI) that follows TBI (Oehmichen et al., 2009). Although DAI has most typically been described in association with rotational forces or severe deceleration (Oehmichen et al., 2009), it appears to also be the major process in this model of blast exposure in rats with body shielding. Progression in severity over the 2-week period covered in this study suggests that some axons injured by the blast exposure may be amenable to therapeutic salvage during the immediate post-blast period (Büki et al., 1999a, 1999b, 2003; Singleton et al., 2001; Smucker et al., 2009). However, the existence of a therapeutic opportunity must be proven in therapeutic trials with agents targeting axonal injury. Although the histopathologic patterns of axonal degeneration suggest a direct effect of the blast waves on CNS axons, it is also possible that special sensory organs such as the eyes and inner ears may also have been affected by the blast exposures. However, microscopic examinations were not performed on those tissues in this study.
Although the noted differences in frequencies of degeneration of Purkinje somas and Purkinje dendrites may relate to the large sizes of the Purkinje neuron dendritic trees in relation to the sizes of the cell bodies of origin (so the Purkinje neuron somas were less frequently seen on section), examination of multiple step sections of cerebellum (at 960-μm intervals) suggested that the Purkinje neuron dendrites were likely to have been more vulnerable to blast injury than the Purkinje neuronal cell bodies. It has been reported by Rossi and associates (2006) that Purkinje neurons exhibit a characteristically weak somal response to axotomy—although the mechanism underlying this finding remains to be determined. Recently, Yue (2007) reported that autophagy in Purkinje axons and axon terminals is robust compared to that seen in cell somas in response to an excitotoxic insult. The possibility that autophagy, or other mechanisms, play a role in this process after blast TBI remains to be explored.
The pattern of granular staining within the molecular layer of the dentate gyrus is most suggestive of synaptic or terminal degeneration. Ultrastructural evaluations would be necessary to clarify the specific site of cellular injury, however. The stratum lacunosum moleculare and the middle to outer portion of the dentate molecular layer (those hippocampal regions primarily affected in these blast-injured rats) represent the terminal points of the perforant pathway (i.e., projecting from the entorhinal cortex to the granule neurons of the dentate gyrus) (Lopes Da Silva et al., 1990), and this represents the primary input pathway from the cortex to the hippocampus. The majority of the rats (but not all) that exhibited this staining pattern within the dentate gyrus also had either neuronal degeneration within the entorhinal cortex, or evidence of axonal degeneration within the entorhinal subcortical zone. Silver staining within the entorhinal cortex and hippocampal dentate gyrus was always more prominent on the right side (contralateral to the blast wave). Although axonal injury in other regions of the brain was frequently bilaterally symmetrical, injury in these other regions was also frequently more severe on the right (contralateral) side. The reasons for greater degrees of brain injury contralateral to the blast exposure are not clear. However, it is appreciated that the air shock front will diffract around the skull in less than 50 μsec, and in this manner may produce a localized imploding shock amplification on the external contralateral side of the head, while the reflected shock pressure at the front is rapidly relieved (Alexander et al., 2010). The timing of this dramatic loading shift coupled with the skull response dynamics may well be the cause of greater contralateral injury, yet be unrelated to impact. It is also possible that “skull flexure” coupled with the diffracting shock front may play a primary role in the distribution of the blast exposure–induced brain lesions (D. Ritzel, personal communication).
We also observed BBB permeability at 24 h after injury; however, this was largely restricted to the cerebral cortex, was not observed in regions with maximal axonal silver staining (such as the brainstem and cerebellum), and was more severe within the cerebral cortex and underlying striatum contralateral to the blast. BBB permeability had resolved by 72 h. This finding adds to our qualitative observation that histopathologic damage overall tended to be greater within brain regions contralateral to the blast exposure, and suggests potentially important regional differences in the pathophysiologic processes that occur in the brain after blast TBI, despite what is generally viewed as a diffuse exposure. Greater disruption of the BBB contralateral to the blast exposure may not be surprising, because it is quite possible that the abrupt underpressure and severe gradients might damage the BBB on the contralateral side, as discussed above. Computational modeling has predicted this possibility (Alexander et al., 2010), although such predictions are highly dependent on the details of the model. Ultimately the BBB is a delicate yet permeable structure of cells maintaining a pressure/material differential between blood and CSF, and hence would be expected to be inherently vulnerable to such severe transient pressure conditions (Kaur et al., 1996). However, we recognize that we cannot completely exclude the possibility of restraint-induced injury artifact from the leather sling, although we specifically designed the restraint as a compliant sling rather than a hard surface to minimize any impact. We also recognize that IgG is a large molecule and more sensitive tracers such as aminoisobutyric acid or gadolinium might demonstrate a more dramatically altered BBB permeability. The anatomical disconnect between regions of maximal axonal injury and BBB disruption was surprising. Our assessment of BBB permeability included a more restricted number of rats than were used for neuropathology and is thus exploratory. Further studies examining the relationship between BBB injury and axonal injury using more sensitive tools to assess BBB damage are needed. The presence of BBB damage in the cortex, however, could have relevance given the prominent acute brain swelling often seen in severe blast TBI in combat casualty care (Ling et al., 2009). Further studies are indicated.
These histopathologic findings build on the work of Long and associates (2009), and further suggest that rodents can be used to appropriately model blast injury. They also build on the seminal work of Hall and colleagues (2008), that showed a surprising amount of diffuse fiber tract degeneration (using silver staining) in the controlled cortical impact model. Our findings also corroborate those of recent work by Svetlov and co-workers (2010), who reported silver staining in deep brain structures in rats after blast exposure (52 PSI, 10 msec), although a rather different model was used with the rat positioned outside of the shock tube. Whether all silver-stained axons are irreversibly damaged is not known. Nevertheless, the ability of silver degeneration stains to highlight degenerative processes has been well validated at the ultrastructural level (Heimer and Peters, 1968; Ikonomidou et al., 2000). In the present study, the amino cupric silver stain was the most sensitive stain used for delineating fiber tract injury, especially in relation to patterns of connectivity between brain regions. Our findings of minimal staining by APP after blast TBI in rats are similar to those of Risling and associates (2009), who reported no APP immunostaining in rats after exposure to an explosive blast (20–34 PSI) in a shock tube despite functional impairment and alterations in gene expression on gene array. This contrasted with the robust APP staining seen in a rotational angular deceleration model. Our work suggests that cupric silver staining represents an important and underutilized methodology for studying structural outcomes after experimental blast TBI in relation to other diagnostic/prognostic approaches and therapy.
We conclude that a survivable blast exposure in rats with body shielding can result in acute and enduring axonal injury, particularly in the cerebellum and brainstem, and that a peracute increase in BBB permeability occurs in the cortex, despite limited evidence of neuronal death and only a modest glial response. We report that cupric silver staining was more sensitive than conventional stains or APP immunohistochemistry to identify axonal degeneration. However, given the sensitivity of the silver stain, it is essential that the morphologic staining patterns observed confirm axonal injury. Given the similar findings that are emerging in the human condition, future studies of injured brains from rats with longer recovery periods—coupled with functional outcome studies—are needed to further define and characterize the extent of brain injury in this and other blast models, and to define the therapeutic potential of this target in blast TBI. Such an approach should prove useful in studies utilizing both single and repeated blast exposure models.
Footnotes
Acknowledgment and Author Disclosure Statement
This work was supported by the DARPA PREVENT blast program N66001-10-C-2124. The views, opinions, and/or findings contained in this manuscript should not be interpreted as representing the official views or policies, either expressed or implied, of DARPA or the Department of Defense. Dr. Switzer has commercial interests in a company (NeuroScience Associates) that performs CNS amino cupric silver stains for academia, government, and industry.