
Abstract
Traumatic brain injury (TBI) increases brain beta-amyloid (Aβ) in humans and animals. Although the role of Aβ in the injury cascade is unknown, multiple preclinical studies have demonstrated a correlation between reduced Aβ and improved outcome. Therefore, therapeutic strategies that enhance Aβ clearance may be beneficial after TBI. Increased levels of ATP-binding cassette A1 (ABCA1) transporters can enhance Aβ clearance through an apolipoprotein E (apoE)-mediated pathway. By measuring Aβ and ABCA1 after experimental TBI in C57BL/6J mice, we found that Aβ peaked early after injury (1–3 days), whereas ABCA1 had a delayed response (beginning at 3 days). As ABCA1 levels increased, Aβ levels returned to baseline levels—consistent with the known role of ABCA1 in Aβ clearance. To test if enhancing ABCA1 levels could block TBI-induced Aβ, we treated TBI mice with the liver X-receptor (LXR) agonist T0901317. Pre- and post-injury treatment increased ABCA1 levels at 24 h post-injury, and reduced the TBI-induced increase in Aβ. This reduction in Aβ was not due to decreased amyloid precursor protein processing, or a shift in the solubility of Aβ, indicating enhanced clearance. T0901317 also limited motor coordination deficits in injured mice and reduced brain lesion volume. These data indicate that activation of LXR can reduce Aβ accumulation after TBI, and is accompanied by improved functional recovery.
Introduction
T
Aβ homeostasis is maintained by balancing production (APP processing) and clearance (Aβ degradation). Clearance of Aβ is positively enhanced by activity of the cholesterol efflux transporter ATP-binding cassette A1 (ABCA1). Pharmacological elevation of ABCA1 levels with liver X-receptor (LXR) agonists decreases Aβ levels in vivo and in vitro (Burns et al., 2006; Koldamova et al., 2005b; Sun et al., 2003). Recently, three independent groups concurrently published studies on the effects of ABCA1 knockout in APP-transgenic mice. Each group found that Aβ deposition was increased in ABCA1 knockout mice, despite no change in APP processing (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Wahrle et al., 2005). Similarly, knocking out LXR enhances Aβ deposition in APP-transgenic mice (Zelcer et al., 2007), again with no appreciable effect on APP processing. Conversely, overexpression of ABCA1 in APP-transgenic mice reduces Aβ deposition (Wahrle et al., 2008). ABCA1 enhances lipidation of apolipoprotein E (apoE). Lipidated apoE binds to Aβ and delivers it to microglia for degradation by neprilysin (Fitz et al., 2010; Jiang et al., 2008). The richer the apoE lipidation, the more Aβ will be delivered for degradation. ABCA1 is the primary factor in the brain that regulates apoE lipidation (Fitz et al., 2010; Jiang et al., 2008).
As elevated ABCA1 enhances Aβ clearance, we hypothesized that increasing ABCA1 could prevent the TBI-induced increase in Aβ. To test this we administered an LXR agonist prior to, and following, experimental TBI, and examined Aβ, ABCA1, inflammatory markers, functional outcome, and lesion volume.
Methods
Controlled cortical impact injury
All procedures were carried out in accordance with protocols approved by the Georgetown University Animal Care and Use Committee. The controlled cortical impact (CCI)-injury device was designed and built at Georgetown University, and consists of a microprocessor-controlled pneumatic impactor with a 3.5-mm-diameter tip. Moderate injury was induced by an impactor velocity of 6 m/sec and deformation depth of 2 mm, as previously described (Loane et al., 2009). C57BL/6J mice (23–25 g) were anesthetized with isoflurane (induction at 4% and maintenance at 2%) in a gas mixture containing 30% oxygen/70% nitrous oxide, and administered through a nose mask. Depth of anesthesia was assessed by monitoring respiration rate and pedal withdrawal reflexes. The mouse was placed on a heated pad, and core body temperature was maintained at 37°C. The head was mounted in a stereotaxic frame, and the surgical site was shaved and cleaned with chlorhexidine diacetate and ethanol scrubs. A 10-mm midline incision was made over the skull, the skin and fascia were reflected, and a 4-mm craniotomy was made on the central aspect of the left parietal bone. The impounder tip of the injury device was then extended to its full stroke distance (44 mm), positioned on the surface of the exposed dura, and reset to impact the cortical surface. After injury, the incision was closed with interrupted 6-0 silk sutures, anesthesia was terminated, and the animal was placed in a heated cage to maintain normal core temperature for 45 min post-injury. All animals were monitored carefully for at least 4 h after surgery, and then daily thereafter. Sham animals underwent the same procedure as injured mice, except they did not receive an impact.
Drug administration
The LXR agonist T0901317 (Cayman Chemicals, Ann Arbor, MI) was suspended in 0.5% methylcellulose (Sigma-Aldrich, St. Louis, MO) with 2% Tween-20 (Sigma-Aldrich). The animals received 25 mg/kg administered once daily by oral gavage at a final volume of 5 mL/kg.
Study 1
T0901317 was administered using a pretreatment dosing regimen. Mice were administered either vehicle or T0901317 for 3 days prior to injury, then at 15 min post-injury.
Study 2
T0901317 was administered as a pretreatment as described for study 1, and compared to a post-injury treatment dosing regimen. The post-treatment mice received vehicle for 3 days prior to injury, and either T0901317 or vehicle 15 min post-injury.
Study 3
For behavioral and lesion volume analysis at 7 days post-injury, at 3 days post-treatment mice received either T0901317 or vehicle, and were treated as in study 2. They continued to receive daily administrations of T0901317 until day 7.
The timing of the drug dosing regimens was based on our previous work with T0901317 (Burns et al., 2006; Eckert et al., 2007; Hoe et al., 2007). We have shown that a single oral dose of T0901317 is sufficient to cause changes in ABCA1 at 24 h after injection (Hoe et al., 2007); however, we also included a pretreatment regimen as a proof-of-concept study to ensure full penetration and priming of ABCA1 prior to TBI induction and Aβ production. For study 3, we administered T0901317 on a daily basis to ensure that ABCA1 remained elevated throughout the critical period of Aβ elevation (1–3 days). We have previously shown that daily treatment with T0901317 for 7 days maintains elevated LXR-responsive proteins, including ABCA1 (Burns et al., 2006; Eckert et al., 2007).
Tissue homogenization
The ipsilateral cortex was homogenized in 10 volumes of ice-cold tissue homogenization buffer containing 250 mM sucrose, 20 mM Tris-base, 1 mM EDTA, and 1 mM EGTA (pH 7.4), with mammalian tissue protease inhibitor cocktail. The homogenate was mixed 1:1 with 0.4% diethylamine (DEA) and 100 mM NaCl solution using a ground glass pestle in a dounce homogenizer. This mixture was centrifuged at 135,000g for 1 h. The supernatant was removed (DEA-soluble fraction) and neutralized with 10% 0.5 M Tris-HCl (pH 6.8). The pellet was resuspended in RIPA buffer containing 50 mM Tris-HCl, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA, and protease inhibitor cocktail (pH 7.4) (all chemicals were from Sigma-Aldrich), sonicated for 10 sec, and the supernatant was removed (RIPA-soluble fraction) and frozen until needed for Western blotting. The remaining pellet was resuspended in 1 volume of 70% formic acid, sonicated, and centrifuged at 135,000g for 1 h at 4°C. The supernatant was neutralized in 20 volumes of neutralization buffer containing 1 M Tris-base and 0.5 M Na2PO4.
Aβ and IL-1β enzyme-linked immunosorbent assay
Aβ levels were quantified in the DEA and FA soluble fractions using a commercially available ELISA kit (Wako Chemicals, Richmond, VA), which uses three antibodies to specifically detect Aβ40 and Aβ42 (BAN50/BA27 and BNT77/BA27), as first described by the Younkin laboratory (Suzuki et al., 1994). Aβ levels were measured according to the manufacturer's instructions.
IL-1β was measured from the DEA soluble fraction using a commercially available IL-1β ELISA (R&D Systems, Inc., Minneapolis, MN), according to the manufacturer's instructions. All readings were converted to picomoles per milligram protein.
Western immunoblot analysis
Protein levels were measured using the RIPA soluble fraction. The protein concentration was determined by BCA assay (Pierce, Rockford, IL), and the samples were equalized. Protein samples were resolved by either 7%, 10%, or 4–12% SDS-PAGE gel, transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA), and blocked for a minimum of 1 h in blocking buffer (5% skim milk in PBS containing 0.05% Tween 20 [PBS-T]). The membranes were incubated overnight at 4°C with antibodies for LDLR (1:1000, a kind gift from Dr. Guojon Bu, Washington University, St. Louis, MO), ABCA1 (1:1000; Novus, Littleton, CO), full length APP and APP C-terminal fragment (1:1000 APP-CTF, antibody C1/6.1 against the C-terminus of APP, a gift from Dr. Paul Mathews, Nathan S. Kline Institute, Orangeburg, NY), BACE1 (1:1000; Millipore, Billerica, MA), COX-2 (1:1000; Cayman Chemicals), and β-actin (1:10,000; Sigma-Aldrich), in PBS containing 1% skim milk. The membranes were washed (4 × 10 min in PBS), and incubated in the appropriate horseradish peroxidase-conjugated secondary antibodies (anti-mouse IgG or anti-rabbit IgG; 1:3,000, Jackson ImmunoResearch, West Grove, PA) for 1 h at room temperature. The membranes were washed, and protein complexes were visualized using SuperSignal West Dura Extended Duration Substrate (Pierce). Protein bands were quantified by densitometric analysis using QuantityOne Basic software (Bio-Rad). The data presented are expressed as percentages of control.
Motor function evaluation
We evaluated motor recovery of the animals using a modified beam-walking task, a method that is particularly good at discriminating fine motor coordination differences between injured and sham-operated animals (Loane et al., 2009). The modified device consists of a narrow wooden beam 8 mm wide and 120 mm long, which is suspended 300 mm above a tabletop. The mouse was placed on one end of the beam and the number of footfaults for the right hindlimb was recorded over 50 steps. No training periods were performed, and the first trial was performed 1 day after CCI surgery, and again on days 3, 5, and 7. All studies were performed double-blind.
Magnetic resonance imaging
T2-weighted magnetic resonance imaging (MRI) at 7 days was used to measure lesion volume following TBI, as previously detailed elsewhere (Faden et al., 2003). Lesion volume analysis by MRI closely correlates with that calculated from histological sections (Faden et al., 2003). At 7 days after TBI all animals were anesthetized using isoflurane (induction at 4% and maintenance at 1.5%) in a gas mixture containing 30% oxygen/70% nitrous oxide applied through a nose-mask, and subjected to MRI using a Bruker 7T/21 cm Biospec-Avance system (Bruker, Ettingen, Germany). Briefly, the animals were placed in an acrylic glass animal bed with a heating pad warmed to 37°C to maintain body temperature. Respiratory gating to reduce motion artifacts was achieved using a respiratory monitor. The animal bed was positioned so the animal's head was in the center of the magnet within a 72-mm 1H birdcage resonator (Bruker). Field homogeneity across the brain was optimized and a sagittal scout image acquired (RARE image, FOV = 4 × 4 cm, 128 × 128 resolution, TR/TE = 1500/10 msec with a rare factor of 8, making the effective TE = 40 msec). Multi-slice T2-weighted images were then acquired to obtain 16 contiguous slices commencing at the end of the olfactory bulb and working caudally (FOV = 2.5 × 2.5 cm, slice thickness = 0.75 mm, 256 × 256 resolution, TR/TE = 1500/20 msec, four echo images, and four averages). Lesion volume was estimated from the summation of the damaged areas in each slice (including hypersensitivity), multiplied by slice thickness (ImageJ analysis software, National Institutes of Health). Average lesion volume in cubic millimeters was calculated for each treatment group.
Statistical analysis
Data obtained from independent measurements are presented as the mean ± SE, and analyzed using Student's t-test or analysis of variance (ANOVA), followed by the post-hoc Newman-Keuls multiple comparison test. For the beam-walk test we used a two-way ANOVA with repeated measures, followed by a Bonferroni post-hoc test. All statistical tests were performed using GraphPad Prism software, version 3.02 for Windows (GraphPad Software, Inc., San Diego, CA). Differences were considered significant when p < 0.05.
Results
TBI increases endogenous Aβ40 and Aβ42 levels in non-transgenic C57BL/6 mice
Studies in APP-transgenic mice and other animal models have shown that Aβ increases following experimental TBI (Iwata et al., 2002; Stone et al., 2002; Uryu et al., 2002), and we have recently shown, using non-transgenic mice, that endogenous mouse Aβ40 is also increased following CCI (Loane et al., 2009). Here, we measured levels of both endogenous mouse Aβ40 and Aβ42 in the ipsilateral cortex following TBI. We found that both species of Aβ were increased after brain trauma (Fig. 1A). Aβ40 was increased by 87% at 1 day post-injury (p < 0.05), and remained elevated through 3 days (92% increase; p < 0.05), before returning to baseline levels by 7 days. A similar profile was observed with Aβ42, which was increased by 100% at 1 day (p < 0.01), remained significantly elevated at 3 days (78%; p < 0.05), and returned to baseline by 7 days. These changes in Aβ were accompanied by increases in protein levels of full-length APP (flAPP), and in the production of APP C-terminal fragments (APP-CTF), which are produced following α- and β-secretase cleavage of APP (Fig. 1B). There was a similar increase in protein levels of the β-secretase enzyme 1 (BACE1), as previously reported (Loane et al., 2009; Fig. 1B). These proteins displayed profiles similar to the changes seen in Aβ, peaking at 1–3 days post-trauma.

Traumatic brain injury (TBI) increases amyloid precursor protein (APP) processing, beta-amyloid (Aβ), and ATP-binding cassette A1 (ABCA1) levels in non-transgenic C57BL/6 mice. (
Delayed increase in cortical ABCA1 following TBI
Given the role of ABCA1 in the clearance of Aβ, we wanted to measure the levels of this cholesterol transporter following TBI. We also measured another transporter, the low-density lipoprotein receptor (LDLR), a receptor involved in cellular uptake of cholesterol. We found that LDLR increases in the days immediately following injury (32% increase at day 1, and 70% increase at day 3; p < 0.05), before dropping down below baseline levels at day 7 (28% decrease; Fig. 1D and E). This temporal profile was not shared with ABCA1, however, with protein levels unchanged at 1 day post-injury, but increased by 82% at 3 days (p < 0.01), and 96% at 7 days post-injury (p < 0.01; Fig. 1C).
LXR activation increases ABCA1 protein levels following injury, and prevents the TBI-induced increase in Aβ
Our data show that Aβ levels return to baseline as ABCA1 increases, indicative of the role of ABCA1 in Aβ clearance (Fitz et al., 2010; Jiang et al., 2008). We therefore examined whether increasing ABCA1 levels immediately after trauma could alter the TBI-induced increase in Aβ using T0901317. T0901317 is an LXR agonist that we have previously shown to increase cortical ABCA1 levels within 24 h of a single oral administration (Hoe et al., 2007). We administered a dose of 25 mg/kg, which we have previously shown can increase ABCA1 levels without directly impacting baseline levels of cortical Aβ (Burns et al., 2006). We pretreated mice with T0901317 for 3 days prior to injury, and again 15 min after TBI surgery. Ipsilateral cortical tissue was collected 24 h after injury for biochemical analysis. T0901317 administration caused a significant increase in ABCA1 protein levels in sham-injured mice (p < 0.01), and TBI-injured mice (p < 0.01; Fig. 2A and B). As we saw in Figure 1, TBI also increased Aβ40 and Aβ42 levels at 24 h post-injury. Aβ40 was increased by 46% (p < 0.05), and Aβ42 levels were increased by 42% (p < 0.05; Fig. 2C). However, T0901317 administration attenuated the TBI-induced increase in Aβ (p < 0.05; Fig. 2C), such that Aβ40 or Aβ42 levels were not significantly different between T0901317-treated injured mice and uninjured sham mice. As previously observed in an earlier study, 25 mg/kg T0901317 did not alter Aβ levels in sham-injured mice (Aβ40: 11.98 ± 1.5 fmol/mg protein for sham animals versus 11.42 ± 1.6 fmol/mg protein for T0901317 animals; Aβ42: 3.9 ± 0.3 fmol/mg protein for sham animals versus 4.36 ± 0.6 fmol/mg protein for T0901317 animals; Burns et al., 2006). To test if the reduction of TBI-induced Aβ by T0901317 was due to changes in APP processing, we probed for protein levels of flAPP, APP-CTF, and BACE1. Levels of all proteins were significantly increased at 24 h post-injury (flAPP = 63%, p < 0.05; APP-CTF = 117%, p < 0.01; BACE1 = 74%, p < 0.01), and this was not affected by pretreatment with T0901317 (Fig. 2D–F), suggesting a mechanism other than altered APP processing. Levels of soluble Aβ can also be reduced if there is a shift in Aβ from soluble to insoluble pools (i.e., fibrilization of Aβ). To determine if T0901317 was causing such a shift, we measured levels of insoluble Aβ40 after TBI and T0901317 treatment. Insoluble Aβ40 was barely detectable in sham-injured mice (0.0983 ± 0.019 fmol/g tissue), or in TBI mice at 24 h (0.1123 ± 0.025 fmol/g tissue). T0901317 had no effect on insoluble Aβ40 levels after TBI (0.0886 ± 0.041 fmol/g tissue). These data indicate that T0901317 prevents the accumulation of Aβ following TBI, independent of any effects on APP processing or shifts in solubility.

T0901317 pretreatment prevents the accumulation of beta-amyloid (Aβ) following traumatic brain injury (TBI). (
T0901317 reduces inflammation following TBI
Central inflammatory and immune responses contribute to secondary tissue damage after TBI. The proinflammatory cytokine interleukin-1β (IL-1β) is produced in mice within hours of injury, and is a key promoter of neuroinflammation. IL-1β-mediated neuronal damage does not result from the cytokine itself, but rather from its ability to activate other proinflammatory mediators, such as cyclooxygenase-2 (COX-2). In our model system we measured IL-1β at 1, 3, and 7 days post-trauma, and found that IL-1β was increased by 53% at 1 day after injury (p < 0.05; Fig. 3A), and had returned to baseline levels by day 3. As LXR agonists have been shown to have anti-inflammatory properties (Joseph et al., 2003), we next examined if T0901317 could decrease markers of inflammation in our trauma model. We measured IL-1β in the cytosolic fraction and found that at 24 h post-injury, TBI caused a 40% increase of IL-1β in vehicle-treated mice (p < 0.01). Pretreatment with T0901317 reduced this TBI-induced IL-1β increase, such that levels were indistinguishable from IL-1β levels in sham-treated mice (p < 0.01; Fig. 3B). Similarly, TBI increased COX-2 levels at 1 and 3 days after TBI (p < 0.05; Fig. 3C), and T0901317 pretreatment reduced COX-2 levels at 24 h after injury compared to vehicle-treated mice (p < 0.05; Fig. 3D).
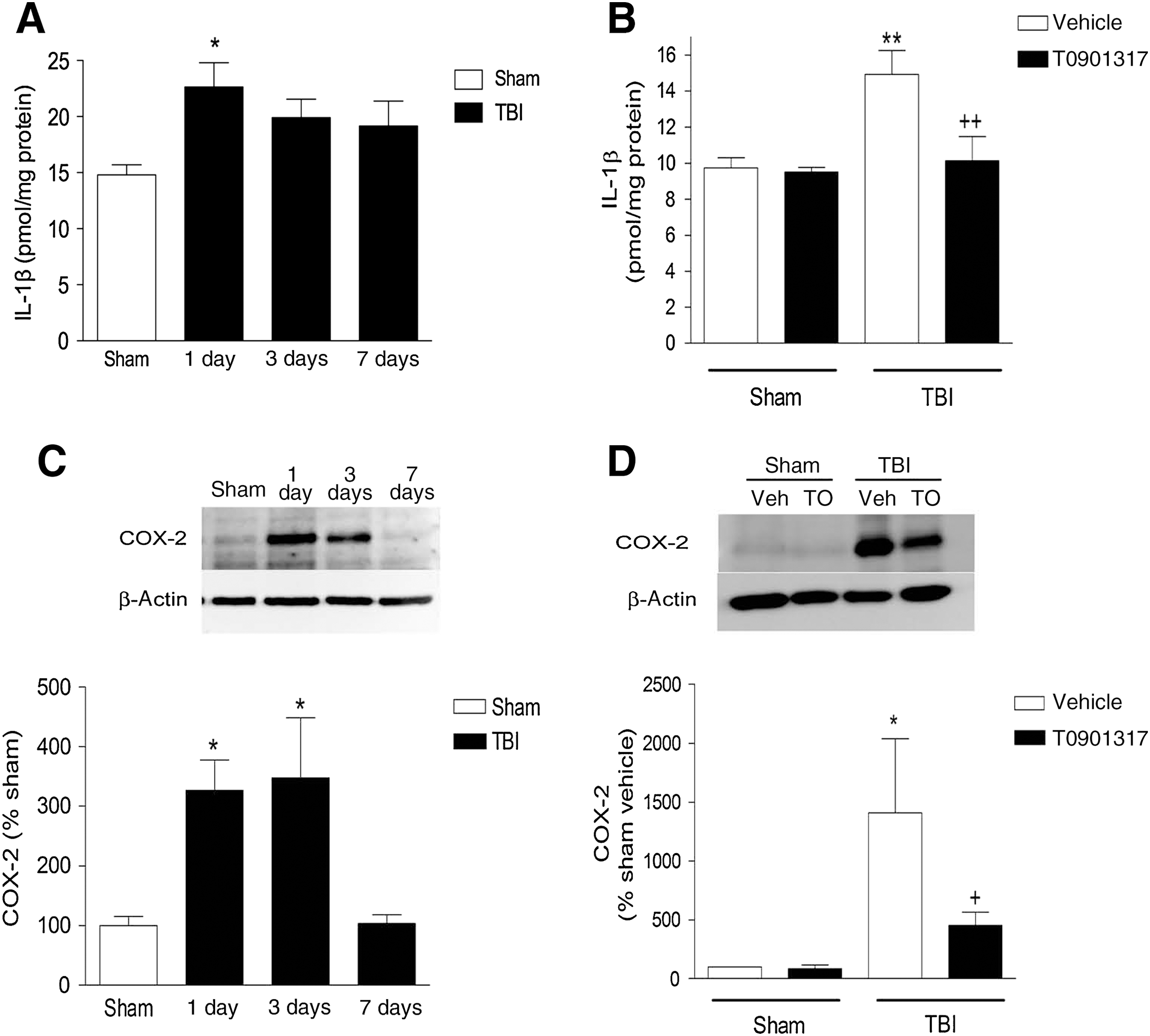
T0901317 pretreatment attenuates inflammation markers following traumatic brain injury (TBI). (
Post-injury administration of T0901317 reduces the Aβ response, but not the IL-1β response, after TBI
We have shown that a 3-day pretreatment with T0901317 can modulate cholesterol transport protein expression, reduce Aβ levels, and attenuate the production of proinflammatory mediators following CCI in mice. In order to ascertain if T0901317 would make a plausible treatment for TBI, we performed a second drug treatment study that repeated our above experiments, but in addition to a 3-day pretreatment group, we also included a post-injury treatment group. For the post-injury treatment group we treated the mice with vehicle for 3 days prior to injury, and T0901317 or vehicle was administered 15 min after surgery. Mice were euthanized 24 h after surgery for measurement of Aβ40, ABCA1, and IL-1β.
Both pretreatment (p < 0.01) and post-injury treatment (p < 0.05) with T0901317 increased levels of ABCA1 in the ipsilateral cortex of injured mice at 24 h (Fig. 4A). Neither treatment regimen had an effect on LDLR levels (Fig. 4A). We again saw increased Aβ40 levels in TBI mice (175% increase compared to sham animals; p < 0.01; Fig. 4B), with both pre- and post-injury treatment attenuating this increase in Aβ40 by approximately 40% (p < 0.01 for pre-injury treatment; p < 0.05 for post-injury treatment; Fig. 4B). While both treatment schedules had similar effects on ABCA1 and Aβ40 levels, this was not the case when we examined the proinflammatory cytokine IL-1β. Levels of IL-1β were increased after TBI (48% increase compared to sham animals; p < 0.05). While T0901317 pretreatment completely blocked this response (p < 0.05), post-injury treatment had no effect on IL-1β levels after injury (Fig. 4C). We also found that T0901317 had no effect on flAPP or APP-CTF levels after TBI (Fig. 4D).
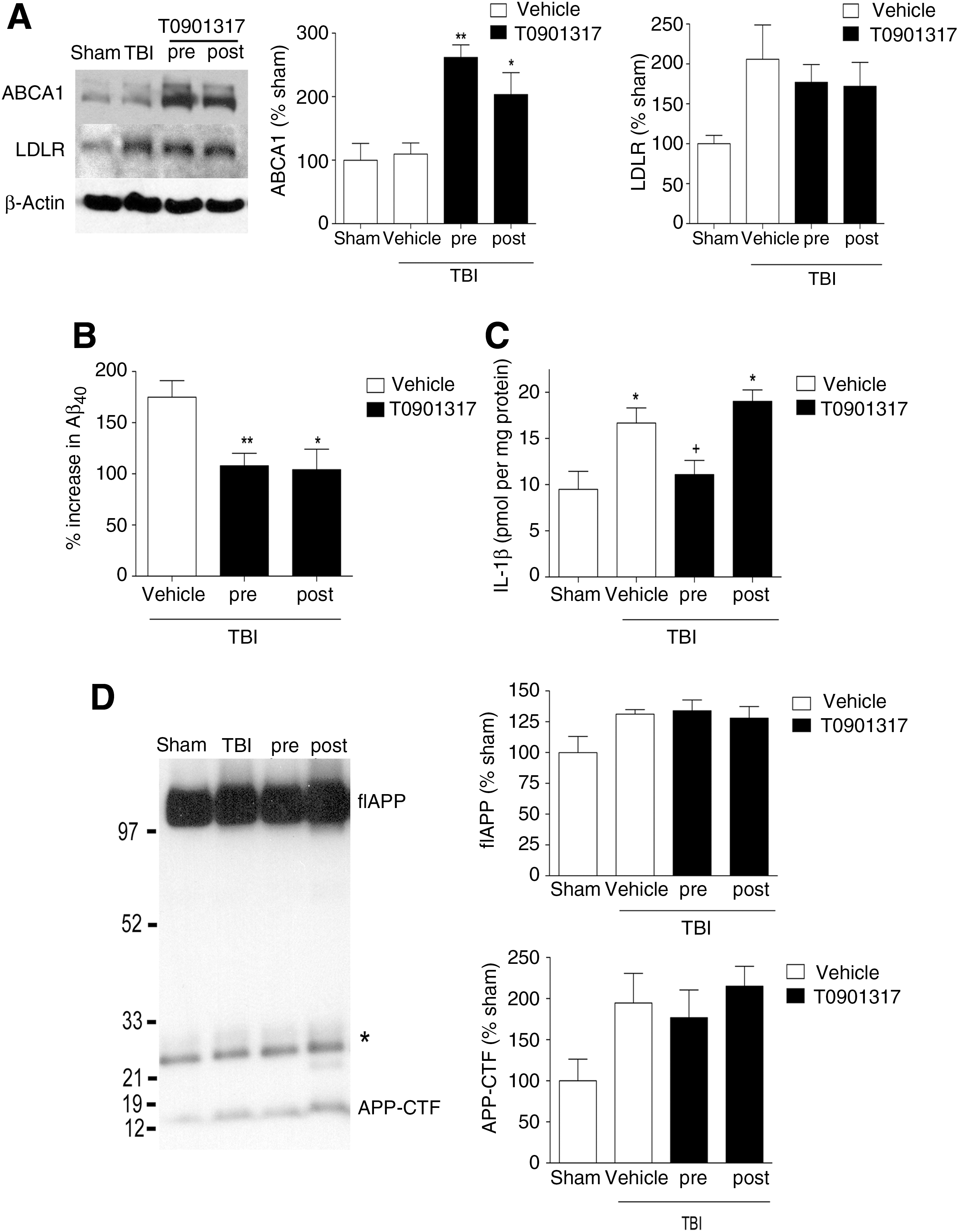
Post-injury administration of T0901317 reduces Aβ, but not IL-1β, following traumatic brain injury (TBI). T0901317 was administered either for 3 days prior to TBI (pre), or as a single injection 15 min after TBI (post). The ipsilateral cortex was collected 24 h after injury. (
T0901317 improves functional recovery and reduces lesion volume in TBI mice
Given T0901317's ability to reduce inflammation and Aβ levels after injury, we wondered if T0901317 could improve TBI-induced behavioral deficits. Beginning with the same pre- and post-injury treatment paradigm as that described above, we continued daily dosing of T0901317 for 7 days after injury, and assessed motor coordination using a beam-walking test. The beam-walking task is a behavioral test that is particularly good at discriminating fine motor coordination differences between injured and sham-operated animals (Loane et al., 2009). Mice were placed on a 8-mm-wide beam and the number of footfaults for the right hindlimb was recorded during 50 steps counted in either direction on the beam. The mice were not trained prior to injury, with the first trial occurring 24 hours after surgery, and testing was repeated on days 3, 5, and 7. Sham-injured mice were able to perform this task at all time points, having < 5 footfaults on each day of testing (Fig. 5A). T0901317 did not alter the ability of sham-injured mice to perform this test. Brain trauma impairs the ability of mice to perform this task, and vehicle-treated TBI mice made 49 ± 1 footfaults the day after injury (p < 0.001 versus sham vehicle animals). Over the 7-day trial period these mice improved slightly, but their performance was still deficient compared to sham-injured mice, with 38 ± 3 footfaults (p < 0.001 versus sham vehicle animals). Pretreatment with T0901317 did not improve performance of injured animals on the day after injury (49.0 ± 0.2 footfaults); however, these mice had significantly fewer faults on day 7 of the trial (22 ± 2 footfaults; p < 0.001 versus vehicle + TBI animals; Fig. 5A), indicating that pretreatment with T0901317 significantly improved recovery of fine motor coordination. Mice treated with T0901317 after injury were also impaired on the day after injury (50 ± 0 footfaults), and these mice showed a trend toward improvement in fine motor coordination over 7 days (30 ± 5 footfaults), but it did not reach statistical significance (Fig. 5A).
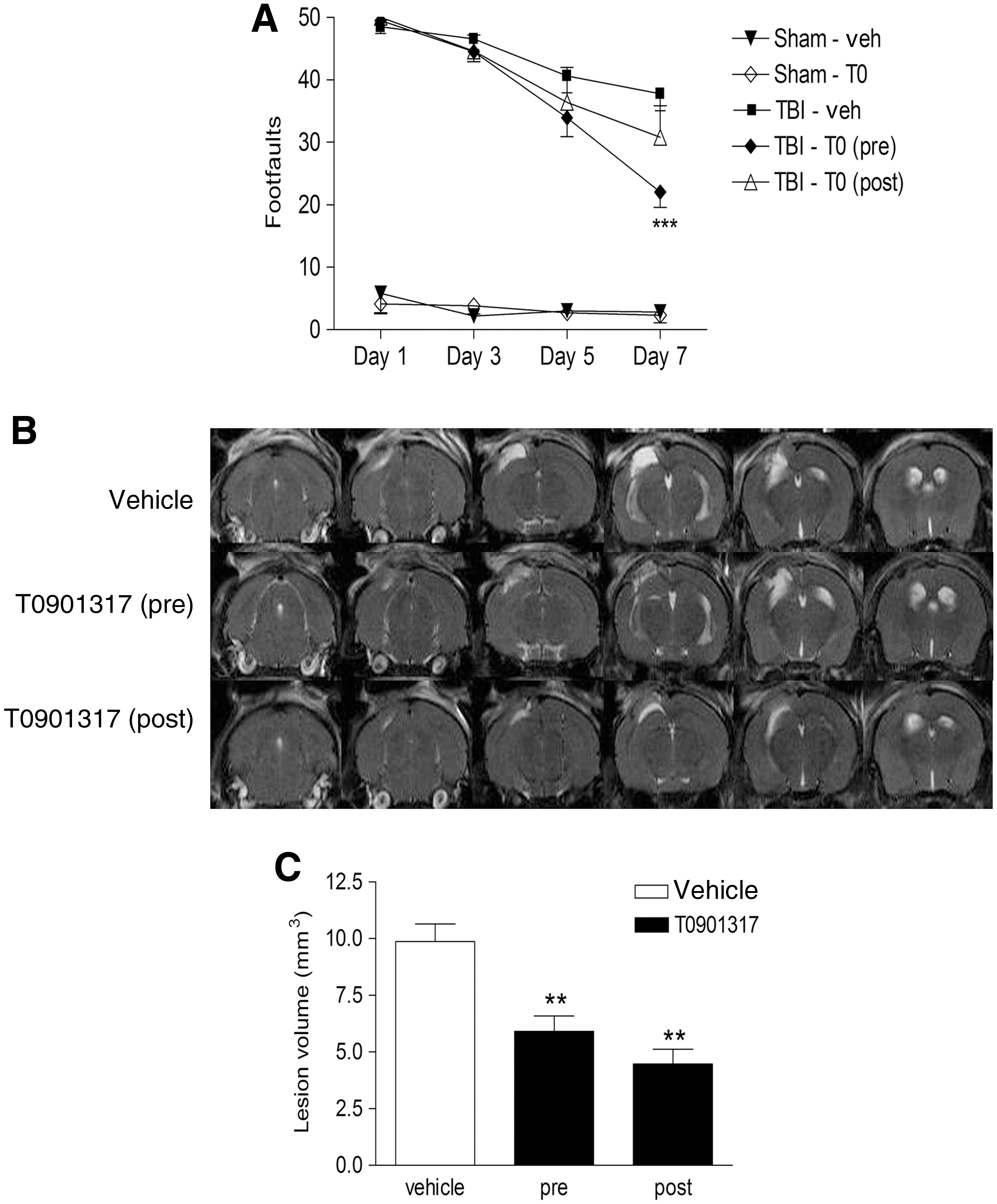
T0901317 attenuates behavioral deficits and reduces lesion volume following traumatic brain injury (TBI). (
Following the final behavioral trial, mice from each treatment group were randomly chosen and imaged using 7-Tesla MRI. Multi-slice, multi-echo T2-weighted images were obtained of contiguous slices through the brain, from the end of the olfactory bulb to the cerebellum. Figure 5B shows representative MRI images from a vehicle-treated mouse, a mouse pretreated with T0901317, and a mouse post-treated with T0901317. The bright areas of hypersensitivity outside of the ventricular areas represent edema and damaged tissue. In T0901317 pretreated mice, the damaged cortical area was smaller than in vehicle-treated mice, and there was a marked decrease in hypersensitivity in the surrounding areas, resulting in a decrease in TBI-induced lesion volume of 40% (p < 0.01; Fig. 5C). Similarly, post-treatment with T0901317 resulted in less damage and edema in the injured cortex, and reduced the TBI-induced lesion volume by 55% (p < 0.01; Fig. 5C).
Discussion
In this study we found that the LXR agonist T0901317 attenuated the TBI-induced increase in Aβ without significantly altering APP processing or Aβ solubility. Rather, it appears that T0901317 enhances clearance of Aβ after TBI. Furthermore, treatment with T0901317 improved functional recovery after TBI and significantly reduced brain lesion volume.
The precise role of Aβ following TBI is unknown. In studies of the injured human brain, performed post-mortem or on survivors, accumulation of Aβ was found in approximately 30% of all patients, and as early as 2 h after injury (Ikonomovic et al., 2004; Roberts et al., 1991). Whether the Aβ increase seen in these cases contributed to neuronal or patient death is unknown. However, the toxicity of Aβ has been well documented. It is implicated as the primary neurotoxic factor in Alzheimer's disease pathogenesis, and is toxic to neurons (Yankner et al., 1989), endothelial cells (Thomas et al., 1996), astrocytes (Brera et al., 2000), and vascular smooth muscle cells (Davis-Salinas et al., 1995). Aβ induces the production of cytokines and reactive oxygen species in microglial cells (Combs et al., 2001), increases sensitivity of primary neurons to excitotoxic damage (Mark et al., 1995; Mattson et al., 1992), and Aβ-mediated cell death demonstrates morphological and molecular features of apoptosis (Imaizumi et al., 1999). Therefore, the increased production of Aβ after brain trauma may be involved in numerous secondary injury cascades that contribute to neurological dysfunction after injury, and preventing Aβ production may be a novel therapeutic strategy for the treatment of TBI.
We have recently reported that targeting the APP secretases after TBI can improve functional recovery, prevent hippocampal neurodegeneration, and reduce lesion volume (Loane et al., 2009). Other reports have also explored the relationship between Aβ and lesion size following TBI in rodents. Abrahamson and colleagues used the pan-caspase inhibitor BAF to prevent caspases-induced cleavage of APP into Aβ following injury (Abrahamson et al., 2006). They successfully reduced Aβ levels 24 h after TBI, and reduced lesion volume 7 days following injury. Another study examined the effects of an apoE-mimetic compound on Aβ and behavior following closed head injury in rodents (Wang et al., 2007). Their drug reduced trauma-induced Aβ42, and significantly ameliorated motor deficits following trauma. Finally, a recent report shows that simvastatin administered 3 h after injury can reduce Aβ levels in an APP-transgenic mouse model (Abrahamson et al., 2009). It should be noted that none of these earlier studies, or the present study, can definitively lead one to conclude that Aβ is mediating secondary injury after TBI, and it may be that any Aβ-lowering effects are secondary to cell survival. However, what can be concluded from these studies is that Aβ is an excellent indicator of functional and histological outcome following TBI.
We examined the profile of APP processing and Aβ accumulation in non-transgenic mice following TBI. We found that the accumulation of Aβ is a transient process in these mice, and that it returns to baseline levels by 7 days post-injury. This is the first profile of both Aβ40 and Aβ42 in non-transgenic mice, and these results may help clarify what has previously been reported in mice overexpressing human APP. CCI in PDAPP mice causes a brief spike in Aβ40 and Aβ42, peaking at 2 h post-injury, and returning to baseline by 6 h (Smith et al., 1998). Longer studies in platelet-derived growth factor promoter expressing APP (PDAPP) mice have shown that CCI can actually decrease deposition of Aβ in the ipsilateral cortex and hippocampus at 4–8 months after injury compared to the uninjured side of the brain (Nakagawa et al., 1999, 2000). Single and repetitive mild CCI injuries in Tg2576 mice caused elevated levels of soluble and insoluble cortical Aβ40 and Aβ42 (Uryu et al., 2002). Finally, studies of APPNLh/NLh mice, a gene-targeted mouse that expresses normal levels of human APP, have elevated Aβ40 only through the first 24 h after CCI, while Aβ42 levels remained elevated through 14 days (Abrahamson et al., 2006). As studies in transgenic mice have yielded variable results, we focused on non-transgenic mice in our studies. However, there are significant differences between rodent Aβ and human Aβ that need to be taken into consideration. Rodent Aβ does not deposit as amyloid plaques in non-transgenic mice, although it is able to form beta-sheet fibrils in vitro (Fraser et al., 1992), albeit not as aggressively as human Aβ can (Boyd-Kimball et al., 2004). Rodent Aβ also differs from human Aβ at 3 amino acid sites: arginine 5, tyrosine 10, and histidine 13 (Yamada et al., 1987). These changes are thought to reduce the ability of rodent Aβ to reduce Cu (II) to Cu (I), and thus rodent Aβ is perceived to lack oxidative stress properties. However, rodent Aβ can still induce protein oxidation and lipid peroxidation in primary neurons, and can trigger apoptosis and cell death (Boyd-Kimball et al., 2004), although at a slower rate than human Aβ. Thus the differences seen in Aβ accumulation after TBI in non-transgenic mice compared to APP transgenic mice and human TBI cases may be due to the reduced ability of mouse Aβ to aggregate as aggressively as human Aβ. As Aβ aggregation has been reported in 30% of human cases, translating treatments that target Aβ from non-transgenic mice to the clinic should be preceded by testing in APP transgenic mouse models.
In the present study, we found that T0901317 reduced Aβ levels in the absence of any changes in APP processing, or any shift of Aβ into the insoluble fraction. These data indicate that T0901317 increases Aβ clearance after TBI. Our findings are supported by multiple studies in APP transgenic mice in which ABCA1 was ablated. These mice had increased Aβ deposition in the absence of altered APP processing (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Wahrle et al., 2005). Conversely, overexpression of ABCA1 in APP transgenic mice reduces Aβ deposition (Wahrle et al., 2008). The mechanism behind the role of ABCA1 in Aβ clearance has recently been shown to be an indirect one, involving apoE as a mediator (Fitz et al., 2010; Jiang et al., 2008). The degradation of Aβ extracellularly (by insulin-degrading enzyme), and intracellularly (in microglia by neprilysin), is greatly enhanced by apoE; however, the ability of apoE to facilitate Aβ degradation is dependent on its lipidation status (Jiang et al., 2008). ABCA1 is essential for the proper lipidation of apoE, and has been shown to mediate the effects of T0901317 on Aβ degradation by microglia (Fitz et al., 2010).
LXR agonists have previously been shown to be neuroprotective in multiple models of neurodegeneration, including spinal cord injury (Paterniti et al., 2010), stroke (Morales et al., 2008; Sironi et al., 2008), and Niemann-Pick type C (NPC) disease (Repa et al., 2007). In spinal cord injury, administration of T0901317 was found to reduce spinal cord inflammation and tissue injury (Paterniti et al., 2010). Following experimental stroke, post-injury administration also reduced the infarct volume, spared cortical tissue, reduced proinflammatory gene expression, and improved neurological scores in rats (Morales et al., 2008). Another group has demonstrated that a single administration of another LXR agonist (GW3965) can reduce lesion volume and improve behavioral outcome (including performance on the balance beam) at 24 and 72 h post-ischemia (Sironi et al., 2008). In NPC mice, administration of T0901317 significantly extended life expectancy, improved motor function, and returned activated microglia to their resting state (Repa et al., 2007). In this article we have examined similar outcome measures, and found improvements in behavioral recovery and reductions in lesion volume. An interesting addendum is that enhanced brain Aβ levels occur in NPC disease, stroke, and spinal cord injury (Burns et al., 2003; Kobayashi et al., 2010; Zhang et al., 2007), but the ameliorating effect of T0901317 on Aβ has not been assessed in any of these models.
In vitro reports have shown that IL-1β can increase mRNA expression and regulate transcription of APP (Forloni et al., 1992; Goldgaber et al., 1989). It is unclear if a similar mechanism is responsible for the increase seen in APP after TBI, but a previous report has demonstrated that the increase in IL-1β occurs prior to the increase in APP protein, suggesting that IL-1β is temporally and spatially available to regulate APP metabolism after trauma (Ciallella et al., 2002). In our study we found that pre-injury treatment with T0901317 significantly attenuated the trauma-induced IL-1β response, without an effect on the trauma-induced APP response. These data suggest that IL-1β does not regulate the production or transcription of APP after injury. We did find that pretreatment with T0901317 successfully inhibited the TBI-induced IL-1β increase in two separate studies; however, post-injury treatment did not. As both treatments successfully reduced Aβ40 levels, it appears that the APP and IL-1β pathways are not necessarily dependent on each other after TBI.
In conclusion, we have shown that TBI increases Aβ40 and Aβ42 in the cortex of injured, non-transgenic mice. Treatment with an LXR agonist enhances ABCA1 levels, and reduces the TBI-induced Aβ peak. These biochemical changes are accompanied by improvements in functional outcome and a reduction in brain lesion volume. These data, along with the effects of T0901317 seen in other models of neurodegeneration, suggest that LXR agonists are neuroprotective in multiple models of CNS dysfunction.
Footnotes
Acknowledgments
This research was supported by grant no. R03NS057635 (to M.P.B.), and funding from the Wright Family through the Memory Disorders Program at Georgetown University (to M.P.B.). We would like to thank Marie Hanscom for technical support; the Small Animal Imaging Laboratory at Georgetown University; Dr. Guojon Bu (Washington University); and Dr. Paul Mathews (Nathan S. Kline Institute) for antibodies; and Dr. Yasuji Matsuoka for helpful comments and discussion.
Author Disclosure Statement
No competing financial interests exist.