
Abstract
Traumatic brain injury (TBI) causes selective neuronal damage in the hippocampus; however, the underlying mechanisms are still unclear. Post-traumatic alterations of ion channel activity, which actively regulate neuronal excitability and thus impact on excitotoxicity, may be involved in TBI-induced neuronal injury. Here we report that hyperpolarization-activated cation current (I h) contributes to the distinct vulnerability of hippocampal neurons in TBI. In a rat model of controlled cortical injury, moderate TBI produced neuronal death of both hippocampal CA3 neurons and mossy cells in the hilus, but not CA1 pyramidal cells. Treatment with lamotrigine, which enhances dendritic I h, ameliorated TBI-induced neuronal damage to CA3 neurons and mossy cells. In contrast, intraventricular administration of I h channel blocker caused cell death in the CA1 region after TBI. Whole-cell recordings revealed that, differently from CA3 neurons, CA1 pyramidal cells expressed larger I h and exhibited a post-traumatic increase of I h amplitude. Moreover, blocking I h led to an increase of neuronal excitability, with greater effects seen in post-traumatic CA1 pyramidal cells than in CA3 neurons. In addition, the I h in mossy cells was dramatically inhibited early after TBI. Our findings indicate that differential changes of I h in hippocampal neurons may be one of the mechanisms of selective cell death, and that an enhancement of functional I h may protect hippocampal neurons against TBI.
Introduction
N
It has been suggested that excitotoxicity is one of the major contributing factors in TBI-induced neuronal injury (Yi and Hazell, 2006). Indeed, several studies have shown that extracellular glutamate concentration is dramatically increased after experimental TBI (Katayama et al., 1990; Palmer et al., 1993). In addition, traumatic injury may cause changes of the properties of ionic glutamate receptors (e.g., an inhibition of the voltage-dependent Mg2+ blockade of NMDA receptors and a reduction of the desensitization of AMPA receptors), leading to an enhancement of glutamate receptor activity (Goforth et al., 1999; Zhang et al., 1996). Moreover, glutamate receptor antagonists have been shown to be neuroprotective against TBI (Bernert and Turski, 1996; Faden et al., 1989; Turski et al., 1998). Consistent with the excitotoxicity hypothesis, a recent study has revealed a significant elevation of intracellular Ca2+ concentration in hippocampal CA3 neurons early after TBI (Sun et al., 2008).
Neuronal excitability, which is dynamically regulated by ion channel activities, may influence excitotoxicity. It is well established that hyperpolarization-activated cation current (I h) plays critical roles in controlling neuronal excitability, by regulating both synaptic inputs and intrinsic membrane properties (Biel et al., 2009; Robinson and Siegelbaum, 2003). Hyperpolarization-activated cation nonselective (HCN1–4) genes encode the channels that underlie I h, a mixed current (Na+ and K+) typically activated by hyperpolarization more negative than −50 mV. Both HCN1 and HCN2 are expressed in the hippocampus, whereas HCN4 is only weakly expressed (Notomi and Shigemoto, 2004). The expression levels of HCN1 in CA1 pyramidal neurons are higher than those in CA3 cells. In contrast, HCN2 is expressed at higher levels in CA3 cells than in CA1 neurons (Monteggia et al., 2000; Santoro et al., 2000). Accumulating evidence indicates that I h activity in hippocampal neurons is sensitive to brain injury, including entorhinal cortex lesions (Brauer et al., 2001) and TBI (Howard et al., 2007). In the present study, we examined the post-traumatic changes of I h in hippocampal neurons, and explored whether I h is involved in TBI-induced neuronal injury in the hippocampus.
Methods
Male Sprague-Dawley rats (220–280 g; Harlan, Indianapolis, IN) were used in the present study. Experimental protocols were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Controlled cortical injury model of moderate TBI
A controlled cortical injury (CCI) model was performed to produce TBI in rats (Dixon et al., 1991). The animals were anesthetized with 2% isoflurane (mixed in 1:2 O2:N2), and placed in a stereotaxic frame. A midline scalp incision was made and a craniotomy (6 mm in diameter) was then performed over the right parietal cortex between the lambda and the bregma and ∼2 mm lateral to the sagittal suture. The dura was exposed and remained intact. An impact device (Benchmark™ Stereotaxic Impactor; MyNeuroLab, St. Louis, MO) attached to a 3-mm impactor tip was used to produce a right fronto-parietal cortex deformation of 2.5 mm, with a velocity of 2.7 m/sec and duration of 0.1 sec. The rats were returned to their cages after recovering from CCI and allowed free access to water and food.
Drug delivery in vivo
A subset of experiments was designed to test the contribution of I h to TBI-induced neuronal injury in the hippocampus. Intraventricular injection (bregma −0.8 mm, lateral 1.5 mm, and depth 3.6 mm) was performed 10 min after CCI to deliver drugs to either block or increase I h pharmacologically. In one group, a specific I h channel blocker, ZD7288 (1 mM, 2.0 μL; Tocris Bioscience, Ellisville, MO) was injected; whereas in another group, lamotrigine (LTG; 2 mM, 2.0 μL; Sigma-Aldrich, St. Louis, MO) was applied to increase dendritic I h (Poolos et al., 2002). In addition, the same amount of saline was used in some rats to serve as a control.
Hippocampal slice preparation and electrophysiological recording
Brain slices were prepared from control animals at 24 h after TBI using procedures similar to those previously described (Deng et al., 2005). Briefly, the animals were anesthetized with ketamine-HCl (80 mg/kg IP) and perfused transcardially with an ice-cold (4°C) sucrose solution containing: 230 mM sucrose, 26 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 10 mM MgSO4, 10 mM glucose, pH 7.4, 290–305 mOsm/L, equilibrated with 95% O2 and 5% CO2. The brains were quickly removed, and transverse hippocampal slices (400 μm) were cut using a vibratome (VT1000S; Leica, Nussloch, Germany) in the sucrose solution. The slices were maintained in an artificial cerebrospinal fluid (aCSF) containing: 130 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose, pH 7.4, 295–305 mOsm/L. The aCSF was continuously equilibrated with 95% O2 and 5% CO2, and slices were incubated for 25 min at 33°C and for at least another 45 min at room temperature (∼24°C) before recording.
Recording electrodes were pulled from borosilicate glass (World Precision Instruments, Sarasota, FL) using a horizontal electrode puller (P-97; Sutter Instruments, Novato, CA). The electrodes had resistances of 2–4 MΩ when filled with an intracellular solution containing: 120 mM KMeSO4, 12 mM KCl, 1 mM MgCl2, 1 mM EGTA, 0.2 mM CaCl2, 10 mM HEPES, 2 mM Mg-ATP, and 0.4 mM Na-GTP, pH 7.4, 295–300 mOsm/L. Oxygenated aCSF was used as bath solution, and the flow rate was adjusted to 2–3 mL/min. Neurons in hippocampal slices were visualized with an infrared-differential interference contrast microscope (BX50WI; Olympus Optical, Tokyo, Japan) and a CCD camera. All recordings were performed at room temperature with an Axopatch 200B amplifier (Molecular Devices, Foster City, CA). After tight-seal (>1 GΩ) formation, the electrode capacitance was compensated. During whole-cell recordings, series resistance (8–15 MΩ) was monitored periodically, and cells with >15% change were excluded from the analysis. Signals were filtered at 2 kHz and digitized at a sampling rate of 5 kHz using a data-acquisition program (Axograph 4.6; Molecular Devices).
Whole-cell voltage-clamp recordings were used to record I h in hippocampal neurons. To evoke I h, a series of hyperpolarizing voltage commands (from −60 to −140 in 10-mV steps, 2 sec) were applied from a holding potential of −50 mV. In addition, CdCl2 (100 μM), 4-aminopyridine (2 mM) and tetraethylammonium (5 mM) were added to bath solution to block voltage-dependent Ca2+ and K+ channels. Tetrodotoxin (0.5 μM) was also used to block Na+ channels. These chemicals were purchased from Sigma-Aldrich. The amplitude of I h was calculated as the current difference between the instantaneous current (measured just after the decay of capacitive transient), and the steady-state current (mean current of 50 msec before the termination of each voltage step). Voltage-dependent activation of I h was determined by normalizing the peak amplitudes of tail currents at −50 mV after various hyperpolarizing voltage commands, and then by fitting the current-voltage relationship with a Boltzmann's function. The time constants of activation were estimated by fitting I h traces evoked by various hyperpolarizing steps with a bi-exponential function.
Current-clamp recordings (fast I-clamp mode) were used to examine neuronal excitability. The resting membrane potentials were read immediately after the establishment of whole-cell configuration. To estimate the input resistance, current pulses (from −150 to 100 in 10-pA steps, 0.8 sec) were applied, and the steady-state voltage responses were measured and plotted against the corresponding current pulses. Then input resistance was obtained from the slope of the voltage-current curve. To examine the contribution of I h to input resistance, ZD7288 (30 μM) was applied in bath solution.
Histological assessment of neuronal injury
Both immunohistochemical staining and hematoxylin and eosin (H&E) staining were used to evaluate TBI-induced neuronal injury in the hippocampus. Specifically, immunohistochemistry with antibodies against glutamate receptor subunit 2/3 (GluR2/3) was performed to label hilar mossy cells (Leranth et al., 1996; Toth et al., 1997), which are vulnerable to TBI (Toth et al., 1997). In addition, H&E staining was used to examine neuronal injury in hippocampal CA1 and CA3 regions. For these experiments, rats in the control and 7 days after CCI groups were anesthetized and perfused through the ascending aorta with phosphate-buffered saline (PBS, 0.01 M, pH 7.4) for about 5 min, followed by 4% paraformaldehyde in PBS for 20–30 min. The brains were removed and post-fixed in 4% paraformaldehyde at 4°C overnight. Coronal sections containing the hippocampus were cut (40 μm) with a vibratome. For each rat two sets of hippocampal sections were collected in PBS for immunohistochemistry and H&E staining, respectively.
For immunohistochemical staining, sections were treated with 0.3% H2O2 in PBS for 30 min. After several rinses in PBS, the sections were blocked with normal serum (1% in PBS containing 0.5% Triton X-100) for 30 min at room temperature, and incubated in a solution containing rabbit anti-GluR2/3 antibodies (2 μg/mL; Millipore, Temecula, CA) for 18 h at 4°C. Thereafter, the sections were incubated in a solution containing biotinylated goat anti-rabbit IgG antibodies (1:200; Vector Laboratories, Burlingame, CA) at room temperature for 1 h, and then with avidin-biotin peroxidase complex (1:100 ABC-Elite; Vector Laboratories) at room temperature for 1 h. Subsequently, the sections were reacted with 0.05% 3-3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) in PBS containing 0.01% H2O2. The sections were then mounted on gelatin-coated slides, and processed for microscopic observation.
Neuronal injury in different hippocampal regions was detected in each group treated with ZD7288, LTG, and saline. Our preliminary data showed that the present CCI model caused no overt damage in CA1 region in most animals; however, pharmacological treatment may cause neuronal injury in the CA1 region after CCI. Thus, the animal numbers with overt CA1 damage were compared between groups treated with different drugs. Indeed, ZD7288 treatment after CCI consistently caused overt damage in the middle 1/3 portion of the CA1 region. We therefore counted survival neurons (in a length of 250 μm) within the damaged segment at three levels (bregma −2.8 mm, −3.6 mm, and −4.3 mm; 2 sections at each level, and 6 sections with H&E staining for each rat). The averaged cell number was then compared to that obtained from corresponding CA1 segments of saline-treated rats. Similarly, the neuronal injury in the CA3 region was examined by counting survival neurons within the middle segment of CA3 (400 μm in length), and the mean number of each rat was used for comparison. For hilar mossy cells, GluR2/3-positive cells in the hilus were counted (each section was immediately adjacent to that used for H&E staining) and averaged. Then the mean value was compared between groups.
Statistical analysis
The data are presented as mean±standard error of the mean. In electrophysiological experiments, “n” represents the number of recorded neurons, which were obtained from 2–5 rats per group. Statistical differences were detected using the paired or unpaired Student t-test (StatView 5.0; Abacus Concepts, Berkeley, CA) unless otherwise noted. Changes were considered significant when p<0.05.
Results
Selective neuronal injury in the hippocampus after moderate TBI
Neuronal injury in the hippocampus was examined 7 days after TBI. We found that lateral CCI produced a formation of cavity in the cortex at the impact site in all animals (n=11). Consistently, CCI also caused neuronal damage in the hippocampus. As shown in Figure 1, dramatic neuronal loss (>50%) occurred in both the CA3 and hilar regions in the ipsilateral, but not contralateral hippocampus. In addition, neuronal damage was detected in the ipsilateral CA2 region. However, no overt damage was observed in hippocampal CA1 regions. The present CCI model may cause a moderate TBI, which is different from severe TBI (with a 5–6 mm impactor tip) that is capable of producing neuronal degeneration in CA1 regions (Hall et al., 2008; Singleton et al., 2010). These results demonstrate that hippocampal neurons exhibit differential vulnerability, with CA3 pyramidal neurons and hilar cells more sensitive to TBI.
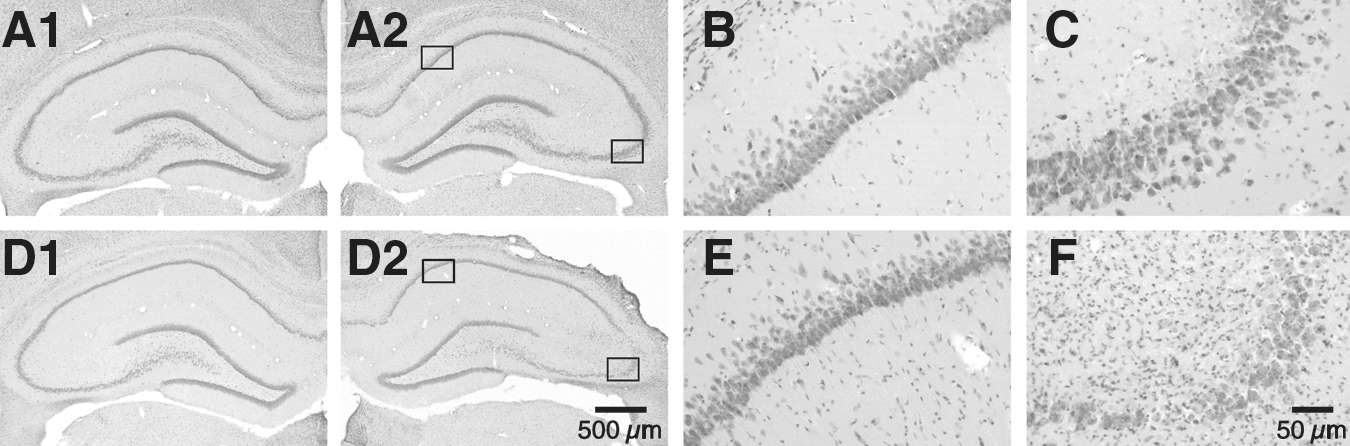
Selective neuronal injury in the hippocampus 7 days after controlled cortical injury (CCI). (
Post-traumatic alterations of Ih in hippocampal neurons
Changes of neuronal excitability early after TBI may influence excitotoxicity and thus neuronal injury. Functional I h is important for the maintenance of intrinsic membrane excitability and the integration of synaptic inputs (Biel et al., 2009; Robinson and Siegelbaum, 2003). We therefore examined I h change in hippocampal neurons 24 h after CCI.
In CA1 pyramidal neurons, CCI caused a significant increase of I h (Fig. 2A and B). For example, when evoked at −90 mV, I h amplitudes were −88.4±3.5 pA and −118.5±11.5 pA in control and post-traumatic neurons (n=6 in each group, p<0.05), respectively. The biophysical features of I h were also compared between control and post-traumatic CA1 neurons. However, no significant change was detected in the kinetics of I h activation (Fig. 2C). With a bi-exponential fitting, it was revealed that the time constant of I h activation was composed of fast and slow components. At a command voltage of −90 mV, the fast time constants were 81.1±6.9 msec in control (n=6) and 77.9±7.3 msec in post-traumatic neurons (n=6, p>0.05), and the slow time constants were 760.5±68.3 msec and 690.7±72.7 msec before and after CCI (n=6 in each group, p>0.05), respectively. In addition, the voltage dependence of I h activation remained unchanged in post-traumatic neurons, as demonstrated by the similar half-activation voltages (control: −89.5±1.2 mV, n=5; CCI: −89.3±1.7 mV, n=6; p>0.05; Fig. 2D).
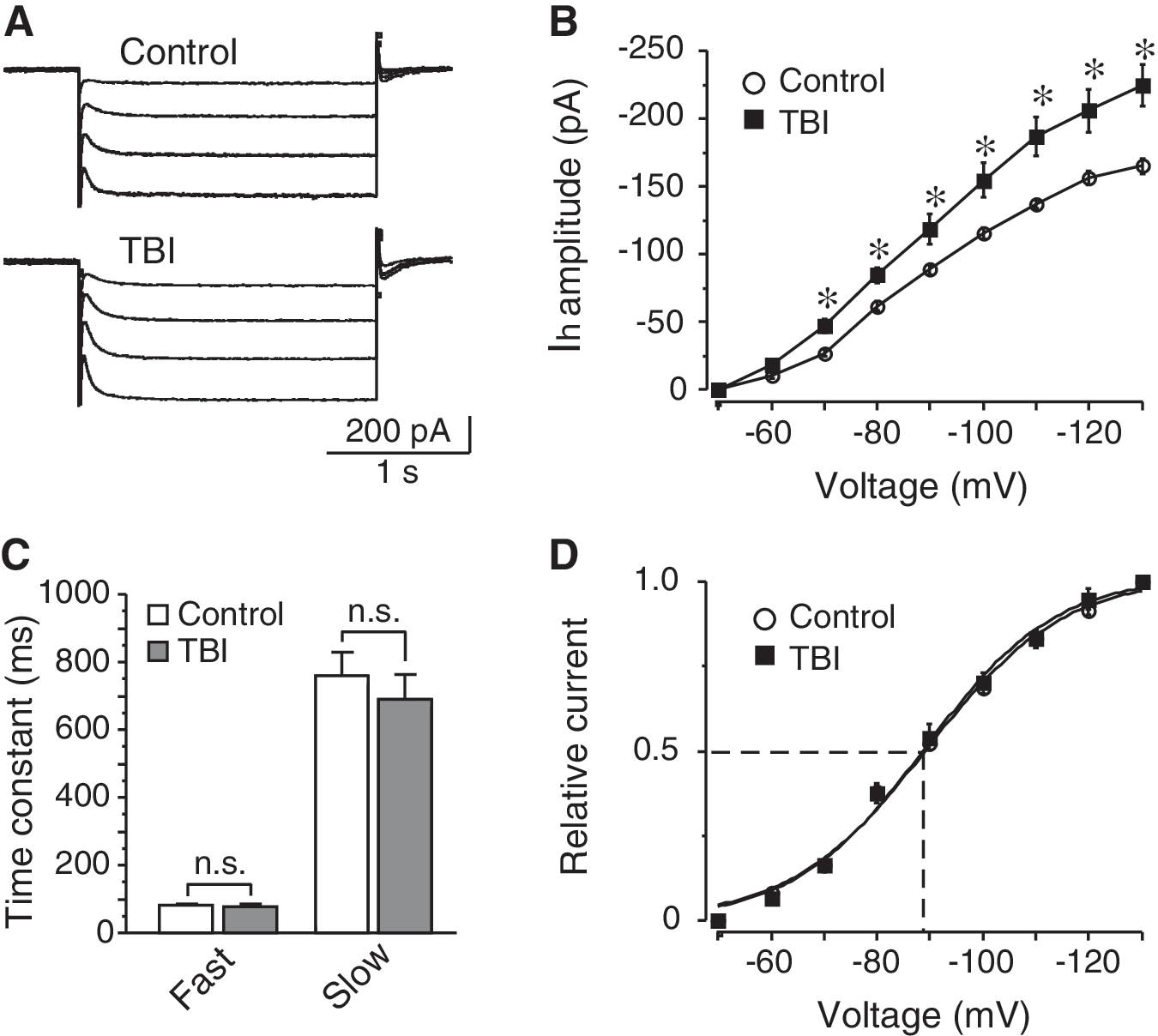
Post-traumatic increase of I
h in CA1 pyramidal neurons. (
Considering that I h in CA1 pyramidal neurons is capable of suppressing neuronal excitability by decreasing input resistance and attenuating excitatory synaptic inputs (Magee, 1998; Poolos et al., 2002), we measured the input resistance of both control and post-traumatic neurons. Indeed, the input resistance of CA1 pyramidal neurons was significantly decreased, from 104.6±11.2 MΩ (n=9) to 73.9±8.4 MΩ 24 h after CCI (n=8; p<0.05, Fig. 3A and C). Bath application of I h channel blocker ZD7288 (30 μM) led to an increase of input resistance, with much larger effects seen in post-traumatic neurons (control: increased to 184.9±19.1 MΩ, by 74.5±8.3%, n=9; CCI: increased to 190.1±20.4 MΩ, by 156.8±13.7%, n=5; p<0.01, Fig. 3C and D). In contrast, the input resistance of CA3 pyramidal cells in the control group (174.4±15.7 MΩ, n=5) was similar to that of post-traumatic CA3 neurons (181.7±17.3 MΩ, n=6; p>0.05; Fig. 3B and C). Moreover, the input resistance in these cells was only slightly increased in the presence of 30 μM ZD7288 (control: increased to 207.5±20.9 MΩ, by 19.0±3.1%, n=5; CCI: increased to 194.7±21.0 MΩ, by 7.7±1.9%, n=6; p>0.05; Fig. 3C and D). These data are consistent with previous studies indicating that CA3 pyramidal cells express little functional I h (Santoro et al., 2000), and indicate that I h in CA3 neurons is not increased after TBI. Thus, CCI-induced I h changes are correlated with the differential vulnerability of hippocampal neurons (i.e., neurons with enhanced I h are relatively insensitive to TBI).
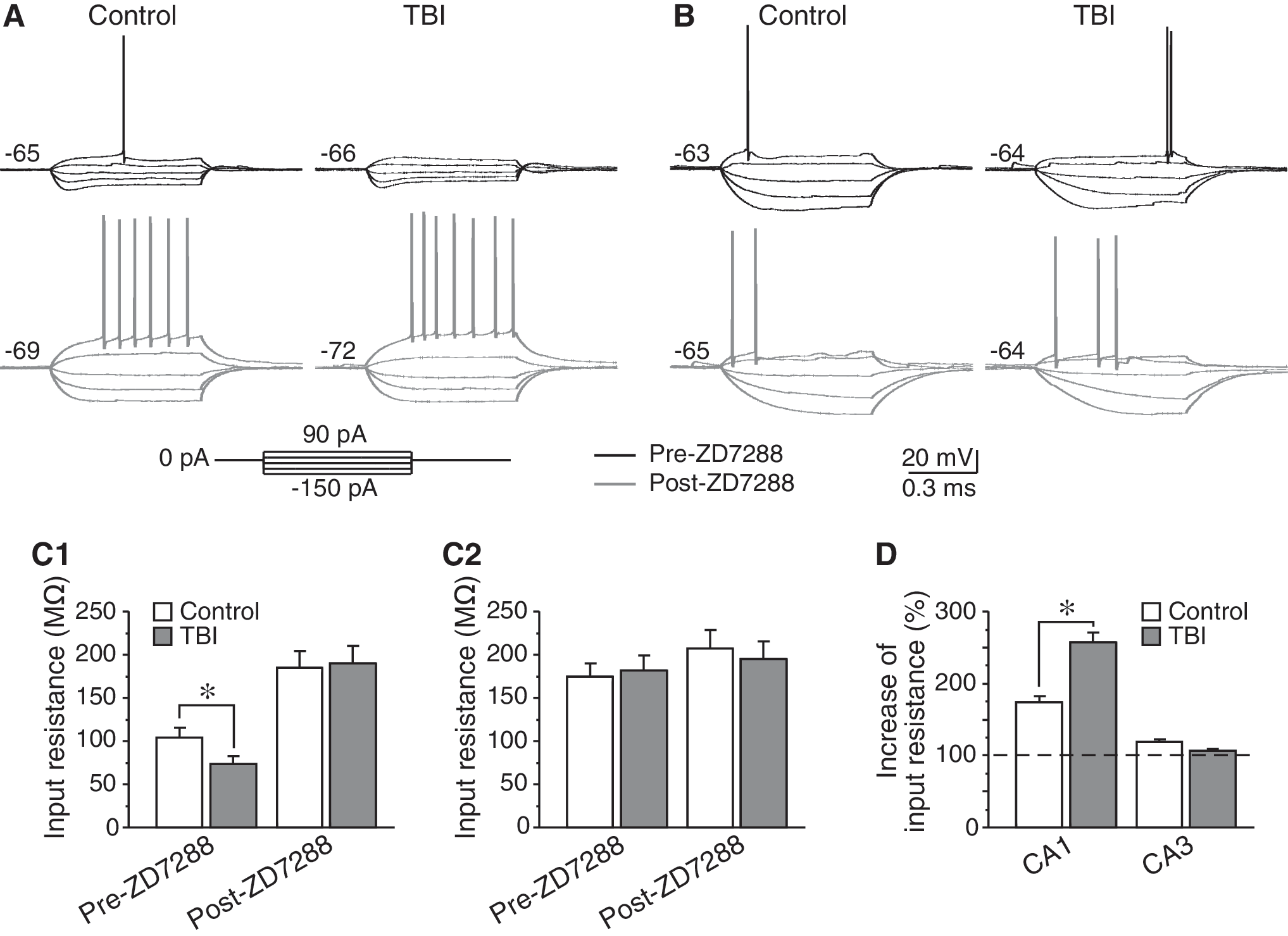
Changes of input resistance in hippocampal pyramidal neurons after controlled cortical injury (CCI). (
Contribution of Ih to post-traumatic neuronal injury in the CA1 and CA3 regions
We further tested the possibility that pharmacological manipulation of I h activity may modulate CCI-induced neuronal injury in the hippocampus. In rats with ZD7288 treatment, CA1 neurons became sensitive to TBI, as CCI caused overt damage in CA1 regions in all animals (n=5). In contrast, only 1 of 6 animals showed overt CA1 region damage in the saline-treatment group (p<0.05 by X 2 test; Fig. 4A,B). Moreover, the neuronal survival in the damaged CA1 region was significantly decreased compared to those in the corresponding regions of saline-treated rats (saline: 143.5±13.9, n=6; ZD7288: 61.8±9.3, n=5; p<0.01; Fig. 4D). Therefore, I h in CA1 pyramidal neurons may be related to the relative resistance of these neurons to TBI. In addition, we found that intraventricular injection of LTG protected CA3 neurons against CCI, and the neuronal survival in CA3 regions was significantly increased (saline: 45.4±6.5, n=6; LTG: 83.2±9.1, n=5; p<0.01; Fig. 4A, C, and E). Together, these findings support the conclusion that in hippocampal pyramidal neurons, I h is neuroprotective against TBI.
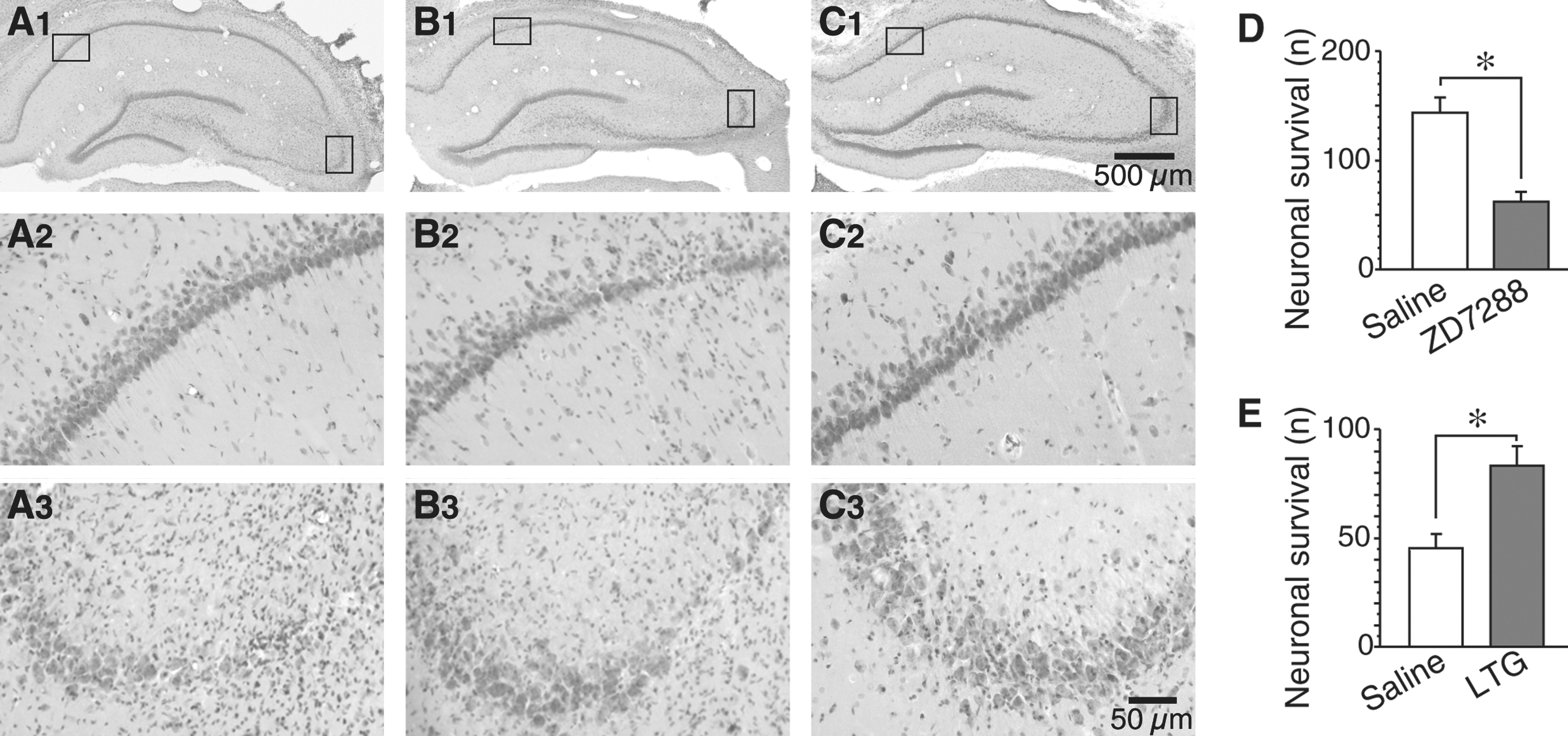
Contribution of I
h to controlled cortical injury (CCI)-induced neuronal injury in the hippocampus. (
Role of Ih in TBI-induced loss of hilar mossy cells
We also investigated the role of I h in hilar mossy cells after CCI, since these neurons possess prominent I h (Brauer et al., 2001; Lubke et al., 1998). It has been shown that mossy cells in the hilus are highly sensitive to TBI (Santhakumar et al., 2000; Toth et al., 1997). We first examined whether I h in mossy cells was altered early after CCI. During recordings, mossy cells were identified based on their electrophysiological characteristics (Brauer et al., 2001; Lubke et al., 1998). Specifically, mossy cells exhibit a small afterhyperpolarization following a spike (∼6 mV, Fig. 5A), which can be easily distinguished from hilar interneurons (>10 mV afterhyperpolarization). Differently from that of CA1 pyramidal neurons, I h in mossy cells was dramatically decreased 24 h after CCI. For example, when evoked at −90 mV, I h amplitudes were −99.2±19.6 pA (n=6) and −43.6±8.0 pA (n=5) in control and post-traumatic cells (p<0.05; Fig. 5B and C), respectively. However, no significant change was detected in the voltage dependence of I h activation (half-activation voltage in controls: −93.5±2.9 mV, n=6; CCI: −92.3±2.6 mV, n=5; p>0.05; Fig. 5D), as well as I h activation kinetics (data not shown). These data suggest that the CCI-induced I h decrease might be involved in the neuronal injury in hilar mossy cells.

Post-traumatic decrease of I
h in hilar mossy cells. (
Then we examined whether upregulation of I h is capable of rescuing mossy cells from injury after TBI. Consistent with previous studies (Toth et al., 1997), lateral CCI caused a dramatic loss of mossy cells (GluR2/3-positive cells) in the ipsilateral hilus. The number of mossy cells was decreased from 30.3±1.5 (n=6) to 10.0±1.5 (n=5; p<0.01; Fig. 6A, B, and D). In animals with LTG treatment, CCI-induced cell loss was significantly reduced (mossy cells: 18.4±1.4, n=5; p<0.01; Fig. 6C and D). Thus our data indicate that an enhancement of I h may protect mossy cells in the hilus against TBI.

Controlled cortical injury (CCI)-induced loss of hilar mossy cells. (
Discussion
The present study demonstrates that CCI causes an increase of I h activity in relatively resistant CA1 pyramidal neurons, which results in a suppression of neuronal excitability. In contrast, TBI-sensitive neurons express little functional I h (e.g., CA3 pyramidal cells), or exhibit decreased I h after CCI (e.g., hilar mossy cells). Consistently, whereas pharmacological blockage of I h exacerbates neuronal damage, an enhancement of I h protects hippocampal neurons against TBI. Therefore, post-traumatic changes of I h may contribute, at least in part, to the differential vulnerability of hippocampal neurons in TBI.
Post-traumatic changes of Ih in hippocampal neurons
Hippocampal neurons express I h, mediated prominently by HCN1 (Notomi and Shigemoto, 2004; Santoro et al, 2000). The functional I h in CA1 pyramidal neurons is much larger than in CA2 or CA3 neurons (Chevaleyre and Siegelbaum, 2010; Santoro et al., 2000). Several lines of evidence indicate that I h in the hippocampus is involved in neurological disorders, such as cerebral ischemia (Fan et al., 2008), epilepsy (Bender et al., 2003; Chen et al., 2001; Shah et al., 2004), and TBI. It has been shown that, after entorhinal cortex lesioning, HCN1 protein expression is downregulated in the CA1 and CA3 regions. Meanwhile, I h activity is inhibited in hilar mossy cells early (1 day) after lesion, accompanied by an increase in the time constant and a negative shift of voltage dependence of I h activation (Brauer et al., 2001). In the present study, we also found a decrease of I h in mossy cells 24 h after CCI. However, I h in CA1 pyramidal neurons was enhanced, suggesting that TBI may cause differential changes of I h in hippocampal neurons. Recently, it was revealed that I h in hilar mossy cells is increased 1 week after fluid percussion injury (Howard et al., 2007). Thus it is possible that TBI may cause a biphasic change of I h in hilar mossy cells (i.e., an initial decrease and a later increase in surviving neurons).
The underlying mechanisms of post-traumatic I h changes in the hippocampus are still unclear. Several intracellular pathways, such as cyclic AMP (Pedarzani and Storm, 1995), Ca2+ (Luthi and McCormick, 1998, 1999; van Welie et al., 2004), and phosphoinositides (Zolles et al., 2006) have been shown to modulate I h in neurons, all of which result in a positive shift of the voltage dependence of I h activation (Biel et al., 2009; Frere et al., 2004). However, we found no change in the half-activation voltage of I h in both CA1 pyramidal neurons and hilar mossy cells after TBI. Our findings suggest that these intracellular pathways might not be involved in the post-traumatic I h changes seen in hippocampal neurons. Alternatively, I h in some neurons is suggested to be regulated by protein kinases, including protein kinase C (PKC), and p38 mitogen-activated protein kinase (p38 MAPK). In hippocampal pyramidal neurons, activation of p38 MAPK causes a positive shift of I h activation and an increase of I h activity (Poolos et al., 2006). However, it has been shown that p38 MAPK remains unchanged in the hippocampus after TBI (Otani et al., 2002). It is therefore unlikely that p38 MAPK contributes to the post-traumatic I h increase in CA1 pyramidal neurons. On the contrary, activation of PKC has been implicated in the inhibition of I h by neuromodulators in other neurons, with or without affecting the voltage dependence of I h activation (Cathala and Paupardin-Tritsch, 1997; Liu et al., 2003). Moreover, PKC activity is significantly increased early (1–3 h) after TBI (Yang et al., 1993). Thus, PKC-mediated inhibition might be associated with the post-traumatic decrease of I h in hilar mossy cells. Nevertheless, it is also possible that, via unknown mechanisms, TBI may cause differential alterations of functional I h channels on cell membranes, leading to post-traumatic changes of I h conductance in hippocampal neurons.
Possible roles of Ih in post-traumatic neuronal injury
Functional I h is an important modulator of neuronal excitability (Biel et al., 2009; Robinson and Siegelbaum, 2003; Santoro and Baram, 2003). Numerous studies suggest that the functions of I h may be inhibitory or excitatory, which is associated with the subcellular distribution and properties of I h channels, the nature and location of synaptic inputs, and the intrinsic membrane activity of particular neurons (Santoro and Baram, 2003). For example, I h may play excitatory roles in striatal cholinergic interneurons, and an inhibition of I h may protect these neurons against cerebral ischemia (Deng et al., 2007, 2008). In contrast, functional I h may play inhibitory roles in hippocampal CA1 pyramidal neurons. Previous studies have revealed a gradient of I h in CA1 neurons, with higher current density in distal dendrites than in soma (Magee, 1998, 1999), which is consistent with morphological findings (i.e., dendritic distribution of HCN1 channels; Lorincz et al., 2002). The dendritic I h might shape the neuronal response to excitatory synaptic inputs by normalizing the distal and proximal excitatory post-synaptic potentials (EPSPs) (Magee, 1999; Williams and Stuart, 2000), and thus influences EPSP summation (Magee, 1999; Nolan et al., 2004). Moreover, a pharmacological increase (with LTG) of dendritic I h shortens the length constant of the distal dendrite, and causes an attenuation of distal EPSPs (Poolos et al., 2002). These results suggest that upregulation of dendritic I h might lead to a depression of neuronal excitability by decreasing input resistance and attenuating excitatory synaptic inputs (Poolos et al., 2002). Considering that neuronal excitability may influence excitotoxicity occurring after TBI, changes of I h activity may be important for TBI-induced neuronal injury.
In the present study, I h in CA1 pyramidal neurons was significantly increased after CCI, which led to a decrease in input resistance in post-traumatic neurons. This observation is consistent with previous studies, that found that hippocampal CA1 neurons were hypoexcitable 24–48 h after fluid percussion injury (D'Ambrosio et al., 1998). Furthermore, intraventricular injection of the I h channel blocker ZD7288 exacerbated CCI-induced neuronal injury in CA1 regions, whereas an increase of I h with LTG treatment attenuated neuronal damage in CA3 regions after TBI. Interestingly, administration of LTG 30 min before fluid percussion injury also reduces neuronal injury in hippocampal CA1–CA3 regions (Hellmich et al., 2007). Therefore, an increase of I h in hippocampal CA1 and CA3 pyramidal neurons may be neuroprotective against TBI. In addition, the present study detected a significant decrease of I h in hilar mossy cells after CCI. It has been shown that mossy cells express HCN1 channels on their dendrites and spines (Brauer et al., 2001). Thus, post-traumatic I h inhibition may cause hyperexcitability in these neurons. Indeed, with LTG treatment, post-traumatic loss of mossy cells was dramatically reduced. The neuroprotective role of I h in hippocampal neurons is also supported by the observation that CA2 neurons, which express much less functional I h than CA1 pyramidal cells (Chevaleyre and Siegelbaum, 2010), were vulnerable to CCI.
In conclusion, our findings indicate that post-traumatic changes in I h may be one of the mechanisms underlying the differential vulnerability of hippocampal neurons to TBI. It is also suggested that increasing I h may be a beneficial intervention for TBI treatment.
Footnotes
Acknowledgments
This work was supported by the Indiana Spinal Cord and Brain Injury Fund (ISDH/A70-0-079212) and American Heart Association grant 0630172N to P.D.
Author Disclosure Statement
No competing financial interests exist.