
Abstract
The development of cytotoxic brain edema resulting in increased intracranial pressure is a major cause of death occurring in the early phase of traumatic brain injury (TBI). Such edema predominantly develops as a consequence of astrocyte swelling. We recently documented that fluid percussion injury (FPI) to cultured astrocytes causes cell swelling. Since aquaporin-4 (AQP4) has been strongly implicated in the development of brain edema/astrocyte swelling in various neurological conditions, this study examined the effect of in vitro trauma on AQP4 protein expression in cultured astrocytes. Exposure of astrocytes to FPI resulted in a significant upregulation of AQP4 protein in the plasma membrane due to neosynthesis, as cycloheximide blocked the trauma-induced AQP4 upregulation. Silencing the aqp4 gene by siRNA resulted in a significant reduction in trauma-induced astrocyte swelling, indicating a critical role of AQP4 in this process. We recently documented that oxidative/nitrative stress (ONS), the mitochondrial permeability transition (mPT), and activation of mitogen-activated protein kinases (MAPKs), contribute to trauma-induced astrocyte swelling in culture. We now show that inhibition of these factors reduces the upregulation of AQP4 following trauma. Since TBI has been shown to activate nuclear factor-kappa B (NF-κB), as well as the Na+,K+,Cl− co-transporter (NKCC), both of which are implicated in brain edema/astrocyte swelling in other conditions, we also examined the effect of BAY 11-7082 and bumetanide, inhibitors of NF-κB and NKCC, respectively, and found that these agents also significantly inhibited the trauma-induced AQP4 upregulation. Our findings show that in vitro trauma upregulates AQP4, and that ONS, MAPKs, mPT, NF-κB, and NKCC are involved in its upregulation.
Introduction
Recent studies from this laboratory have documented that in vitro fluid percussion injury (FPI) results in cell swelling in cultured astrocytes (Jayakumar et al., 2008). Additionally, FPI to cultured astrocytes results in the production of free radicals, the activation of mitogen-activated protein kinases (MAPKs), and in the mitochondrial permeability transition (mPT), as inhibition of these factors led to a significant reduction in trauma-induced astrocyte swelling (Jayakumar et al., 2008). In unpublished observations we also found that FPI to cultured astrocytes results in the activation of the transcriptional factor nuclear factor-kappa B (NF-κB), and that inhibition of such activation resulted in a significant attenuation of trauma-mediated astrocyte swelling.
While the molecular mechanisms involved in astrocyte swelling/cytotoxic brain edema in TBI are not completely understood, a disturbance in osmotic gradients created by altered ion transport systems represents a driving force for the entry of water into astrocytes, ultimately leading to their swelling (Hoffmann and Dunham, 1995). Such water entry has been shown to involve the water channel protein AQP4 (King and Agre, 1996). In the brain, this channel is predominantly expressed in astrocytes, and has been shown to contribute to cytotoxic edema in various neurological conditions (King et al., 2000), including trauma (Vizuete et al., 1999), ischemia (Manley et al., 2000; Taniguchi et al., 2000), neoplasms (Papadopoulos et al., 2004), and hyponatremia (Amiry-Moghaddam et al., 2004). Additionally, mice deficient in AQP4 display a resistance to the development of brain edema in ischemic stroke and in water intoxication (Manley et al., 2000).
Although various studies have documented the upregulation of AQP4 following TBI (Ghabriel et al., 2006; Hu et al., 2005; Sun et al., 2003; Vizuete et al., 1999), potential mechanisms responsible for AQP4 upregulation have thus far not been established. Taking advantage of a simplified in vitro model of FPI, we investigated whether AQP4 is upregulated following trauma to cultured astrocytes, and investigated potential mechanisms for such upregulation. Our findings indicate that trauma to astrocytes causes the upregulation of AQP4, and that inhibitors of oxidative/nitrative stress (ONS), the mPT, MAPKs, NF-κB, and the Na+,K+,Cl− co-transporter (NKCC), all caused a marked attenuation of trauma-induced AQP4 upregulation.
Methods
Astrocyte cultures
Astrocyte cultures were prepared from brains of 1-day-old rat pups by a previously described method (Ducis et al., 1990). Briefly, cerebral cortices were freed of meninges, minced, dissociated by trituration and vortexing, passed through sterile nylon sieves, placed in Dulbecco's modified Eagle medium (DMEM; Life Technologies Corp., Woburn, MA) containing penicillin, streptomycin, and fetal bovine serum, and incubated at 37°C in a humidified incubator in 5% CO2 and 95% air. After 10 days in culture, bovine serum was replaced with 10% horse serum. After 14 days, the cultures were treated and maintained with dibutyryl cAMP (Sigma-Aldrich, St. Louis, MO) in order to enhance cell differentiation (Juurlink and Hertz, 1985). Cultures consisted of at least 98% astrocytes as determined by GFAP and glutamine synthetase immunocytochemistry. The remaining cells consisted of microglia. Experiments were carried out in 3- to 4-week-old cells. Procedures followed guidelines established by the National Institute of Health Guide for the Care and use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee.
In vitro trauma
Trauma was induced in cultured astrocytes using a fluid percussion device first developed by Sullivan and associates (Sullivan et al., 1976), and modified for cell culture (Panickar et al., 2002; Shepard et al., 1991). The injury chamber was coupled to the fluid percussion device with non-distensible Tygon tubing, and the piston was percussed by a weighted pendulum at varying angles of incidence. Five atmospheres of pressure were administered twice for a duration of 25 msec each. Pressures were continuously recorded with a PowerLab system (AD Instruments Inc., Colorado Springs, CO), interfaced with a high-speed pressure transducer. Sham controls were treated exactly like the traumatized cells, except that fluid percussion was omitted.
Inhibitor treatment
The effect of various factor inhibitors, including antioxidants (100 μM PBN, 150 μM vitamin E, 500 U/mL cell permeant catalase, 250 μM L-NAME); cyclosporin A (1 μM), inhibitor of the mPT; UO126 (UO; 10 μM), inhibitor of ERK1/2; SP 600125 (1 μM), inhibitor of JNK and SB 203580 (10 μM), an inhibitor of p38 MAPK, were added to the culture media and the cultures were immediately traumatized. Previously we performed dose-response curves for each inhibitor on trauma-induced astrocyte swelling (Jayakumar et al., 2008), and in the present study we selected the most effective concentration of each inhibitor that showed a significant blockade of astrocyte swelling following trauma. We also recently found that treatment of cultures with BAY 11-7082 (5 μM), an inhibitor of NF-κB activation, as well as bumetanide (100 μM), an inhibitor of NKCC, significantly diminished trauma-induced astrocyte swelling (Jayakumar et al., in press).
Plasma membrane isolation
Plasma membrane enriched fractions were isolated by a previously demonstrated method (Marples et al., 1995). At different time points following trauma, cultures were washed with ice-cold PBS containing protease inhibitor cocktail (PIC), harvested into the same solution, and centrifuged at 4000g (10 min). The pellets were frozen at −80°C for 2 h, subsequently homogenized in 0.2 mL of Tris-EDTA buffer (50 mM, pH 8) containing PIC, and centrifuged at 15,000g (30 min). The pellet was resuspended in the same buffer and centrifuged at 15,000g (30 min), and the final pellet was dissolved in 100 μL RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 1% NP-40, and 5% sodium deoxycholate).
Immunoblotting
Western blot analysis of AQP4 was performed as previously described (Rao et al., 2010). Following normalization of the protein content in each sample, 5–10 μg of each sample in equal volume was carefully loaded in duplicate gels (10% SDS) adjacent to each other (one for AQP4 and the other for α-tubulin), and were simultaneously subjected to electrophoresis under identical conditions. The gels were transblotted onto PVDF membranes. Following transfer, the membranes were probed together using a rabbit affinity-purified AQP4 (1:3000 dilution; Chemicon International, Temecula, CA) and mouse monoclonal α-tubulin (Oncogene, San Diego, CA), and detection was carried out with the ECL system (Amersham Biosciences, Piscataway, NJ). Levels of immunoreactive AQP4 were quantitated with an ImageMaster video documentation system (Amersham Biotech, Uppsala, Sweden). The intensity of each α-tubulin band and the corresponding AQP4 band were quantified, and the ratio of α-tubulin:AQP4 in control was compared with the experimental groups, and the values were expressed as percent change.
Cell volume determination
Cell volume (intracellular water space) was determined using the 3-O-methyl-[3H]-glucose (OMG) method, as previously documented (Kletzien et al., 1975), and modified for astrocyte cultures (Norenberg et al., 1991). In brief, cultured astrocytes were incubated with [3H]OMG (1 mM containing 1 μCi of radioactive OMG; Sigma-Aldrich), and at the end of incubation a small aliquot of medium was saved for specific activity determination. Cultures were washed three times with ice-cold buffer containing 290 mM sucrose, 1 mM Tris-nitrate (pH 7.4), 0.5 mM calcium nitrate, and 0.1 mM phloretin. Cells were harvested into 0.5 mL of 1 N sodium hydroxide. Radioactivity was converted to intracellular water space and expressed as microliters per milligram cell protein. Protein content was determined by the BCA method (Bio-Rad, Hercules, CA).
Transfection of AQP4 siRNA
Transfection of AQP4 siRNA was carried out as previously described (Rao et al., 2010). AQP4 siRNA (ON-TARGETplus SMARTpool), and non-targeting siRNA control (ON-TARGETplus non-targeting Pool), were obtained from Dharmacon Inc. (Lafayette, CO). Transfection of siRNA was carried out using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. In brief, Lipofectamine was diluted 1:50 (v/v) in OPTIMEM low serum medium (Invitrogen) and incubated at room temperature for 5 min. AQP4 siRNA and non-targeting control siRNA (50 nM) were diluted in OPTIMEM medium, added separately to Lipofectamine, and incubated at room temperature for 20 min. Then 0.5 mL of the mixture containing Lipofectamine and siRNA was added to each culture plate containing 2 mL of media, and the transfection was carried out for 48 h. The specific silencing of AQP4 was confirmed by Western blots and by quantitative real-time PCR.
RNA isolation and cDNA synthesis
RNA was isolated from the cultures using the RNAqueous®-4 PCR kit (#AM1914; Ambion, Foster City, CA). RNA was quantified with a spectrophotometer (Nanodrop ND-1000; Thermo Scientific, Pittsburgh, PA). Reverse transcription of RNA to cDNA was done using 2 μg of total RNA with random hexamer primers using the High Capacity cDNA Reverse Transcription Kit (#4368814; Applied Biosystems, Foster City, CA). Only RNA with a 260:280 ratio of 1.9-2.0 was used for cDNA synthesis and PCR analysis.
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) was performed using 10 μL of 1:20 diluted cDNA using the Mx3005P Multiplex Quantitative PCR System (#401513; Stratagene, La Jolla, CA), and qPCR SYBR Green Reagents (Brilliant II SYBR® Green QPCR Master Mix; Agilent Technologies, Santa Clara, CA), with ROX reference dye used as a loading control. Rat-specific primer pair sequences were constructed using NCBI Primer-BLAST. Primer pairs were obtained from Operon Biotechnologies (Huntsville, AL): for ribosomal protein L13a (RPL13a), the forward sequence was 5′-GGCTGAAGCCTACCAGAAAG-3′, and the reverse sequence was 5′-CTTTGCCTTTTCCTTCCGTT-3′. Primer pairs specific for AQP4 (NM_001142366 and NM_012825) were obtained from Qiagen (Valencia, CA) (QuantiTect Primer Assay: Rn_Aqp4_1_SG). qPCR cycling conditions were as follows: an initial 95°C for 10 min, followed by 40 cycles of 95°C for 30 sec, 58°C for 30 sec, and 72°C for 15 sec. MxPro-Mx3005P v4.10 software was used to determine the crossing point for each amplification reaction. The results were exported to Microsoft Excel for analysis. All of the corresponding qPCR data were analyzed using the ΔΔCP method (Pfaffl, 2001), and normalized against one negative control, and the housekeeping gene RPL13a. The fit-point method was used to determine the crossing points for each reaction.
Statistical analysis
Each group consisted of four to five culture dishes per experiment for each time point studied. At least two to four plates were used for Western blot analysis. Experiments were performed in cultures taken from 4–7 separate seedings. Data were subjected to analysis of variance (ANOVA), followed by Tukey's post-hoc comparisons. Intensity unit values obtained from optical density of the bands in Western blots were also subjected to statistical analysis as described above. At each time point, the experimental cultures were compared with their respective sham controls.
Results
In vitro trauma upregulates AQP4 in the plasma membrane
Plasma membrane fractions (PM) were isolated from cultured astrocytes at different time points (30–24 h) after trauma and analyzed for AQP4 protein expression. At 30 min after trauma there was no increase in AQP4 protein, while at 1 h a significant (30%) increase was observed, which further rose to 80% by 3 h, and persisted for up to 6 h, after which there was a gradual decline to approximately 35% at 12–24 h (Fig. 1). We also examined AQP4 levels in the total cellular fraction and observed a similar increase in AQP4 protein expression to that of the PM fraction (data not shown). The pattern of increase in AQP4 expression observed correlated well with our previous observations on the time course of astrocyte swelling following trauma (Jayakumar et al., 2008).
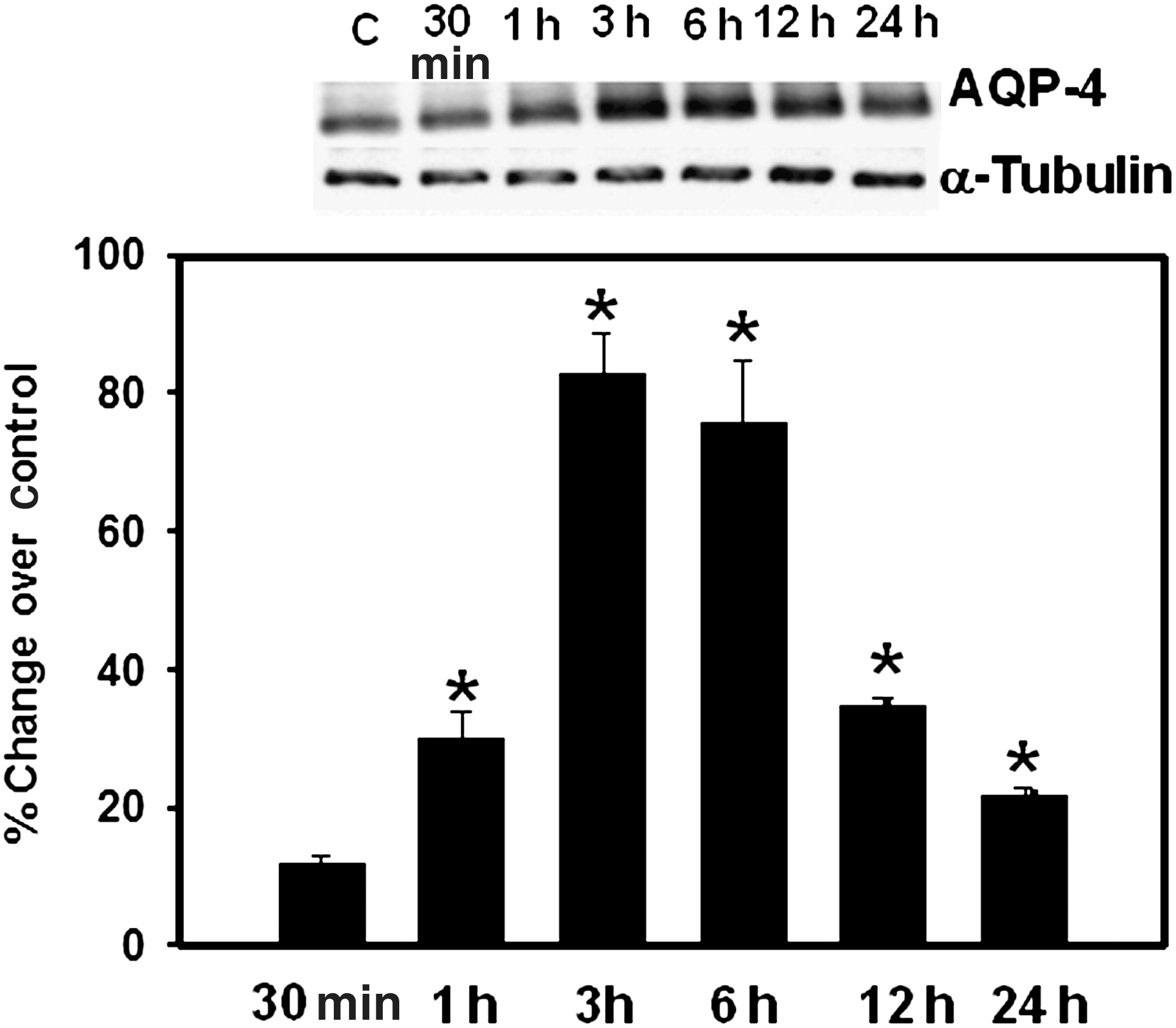
Representative immunoblots showing the time course of aquaporin-4 (AQP4) protein expression in plasma membranes of cultured astrocytes following fluid percussion injury (FPI). AQP4 resolved as a 32-kDa band. The increase in AQP4 in the plasma membrane was first noted at 1 h. Values are mean ± standard error of the mean of three individual plates taken from three separate seedings (*p < 0.05–0.01 versus controls).
Cycloheximide blocks trauma-induced AQP4 upregulation
To examine whether increased AQP4 expression was due to increased stability or increased neosynthesis, cultures were pre-treated (6 h before trauma) with cycloheximide (10 μM), subjected to trauma, and 3 h later (peak time of AQP4 expression), AQP4 protein levels were measured. Cycloheximide significantly (75%, p < 0.01) blocked trauma-induced AQP4 protein upregulation, indicating that neosynthesis is the principal means of the increase in AQP4 expression (Fig. 2).
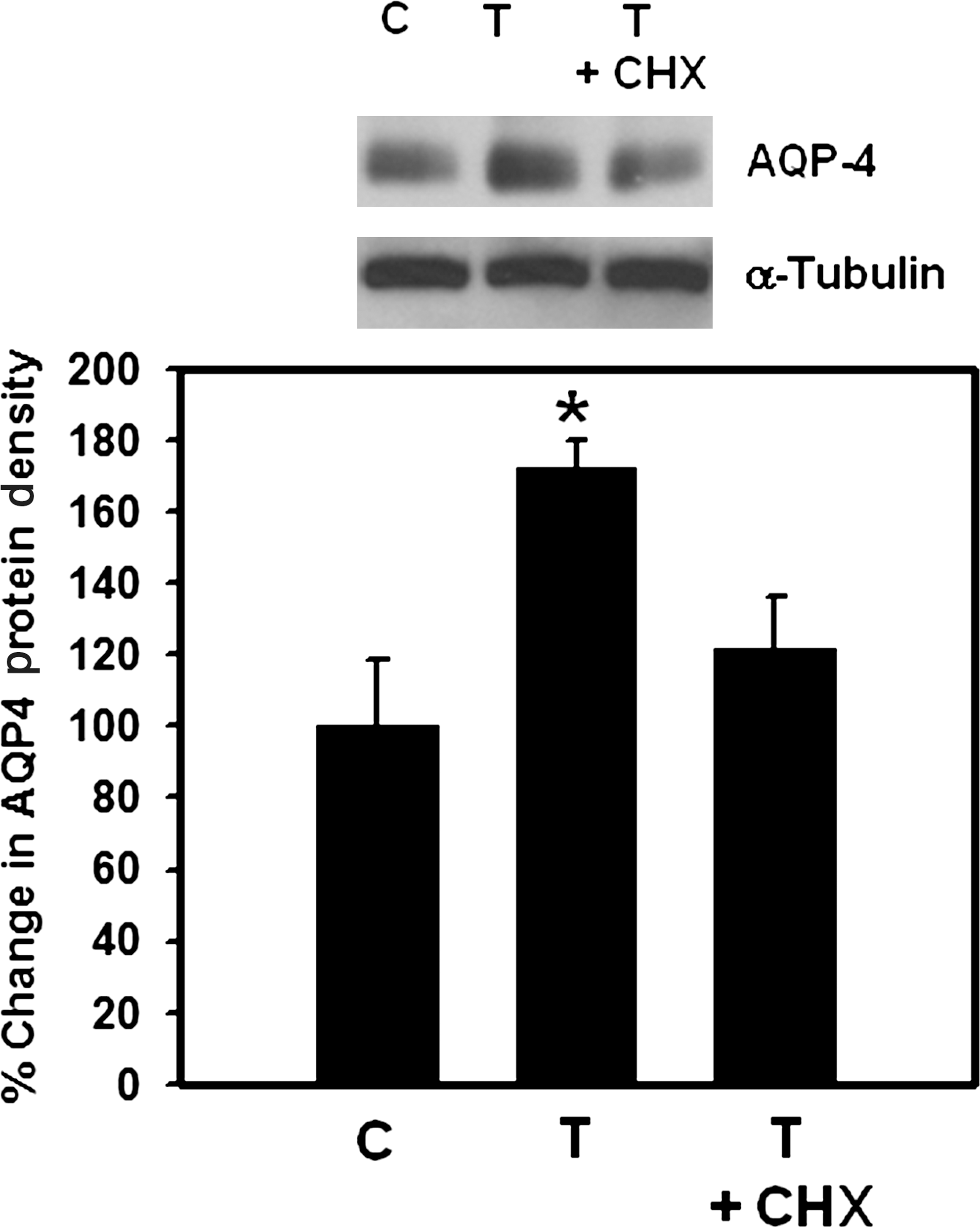
Effect of cycloheximide (CHX) on aquaporin-4 (AQP4) expression in cultured astrocytes following fluid percussion injury (FPI). Cultures were pre-treated (6 h) with CHX, traumatized, and AQP4 expression was examined 3 h after trauma. Values are mean ± standard error of the mean of three individual plates taken from three separate seedings (*p < 0.05–0.01 versus control, C and trauma plus cycloheximide, T +CHX).
AQP4 gene silencing by siRNA blocks trauma-induced astrocyte swelling
To establish the contribution of AQP4 in astrocyte swelling following FPI, astrocyte cultures were transfected with siRNA targeted to AQP4, as previously described (Rama Rao et al., 2010). Such treatment resulted in a 70% reduction in AQP4 protein levels at 48 h after transfection (Fig. 3). We next examined the effect of transfection of cultures with AQP4 siRNA on astrocyte swelling following FPI. Cultures were transfected with AQP4 siRNA, as well as “scrambled” siRNA (control) (50 nM each), and 48 h after transfection, the cells were traumatized and cell volume was determined 3 h following FPI. FPI resulted in significant cell swelling (56 ± 9%) in cultures transfected with control siRNA, whereas cultures transfected with AQP4 siRNA showed a significant reduction (68 ± 5%) in trauma-induced astrocyte swelling (Fig. 4).
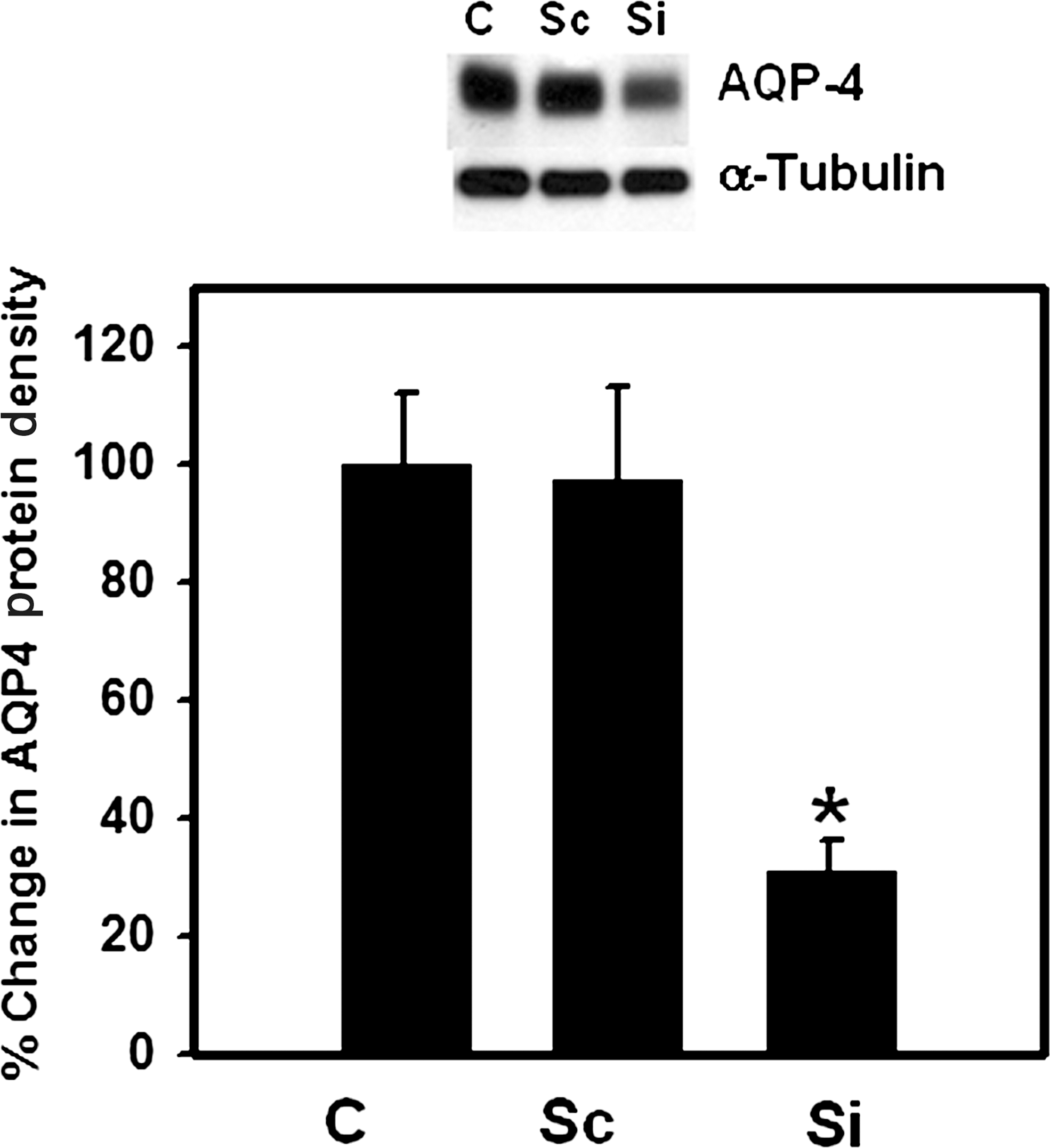
Effect of aquaporin-4 (AQP4) siRNA (50 nM) on AQP4 protein levels. Immunoblots show a significant reduction of AQP4 protein content in cultures transfected with AQP4 siRNA (Si), compared to non-transfected control cells (C). Cultures transfected with non-targeting siRNA (scrambled, Sc, 50 nM) showed no reduction in AQP4 protein. Quantification of AQP4 protein bands show a 65% reduction in AQP4 protein in cultures transfected with AQP4 siRNA. Values are mean ± standard error of the mean of two individual plates taken from two separate seedings (*p < 0.01 versus Sc and C).
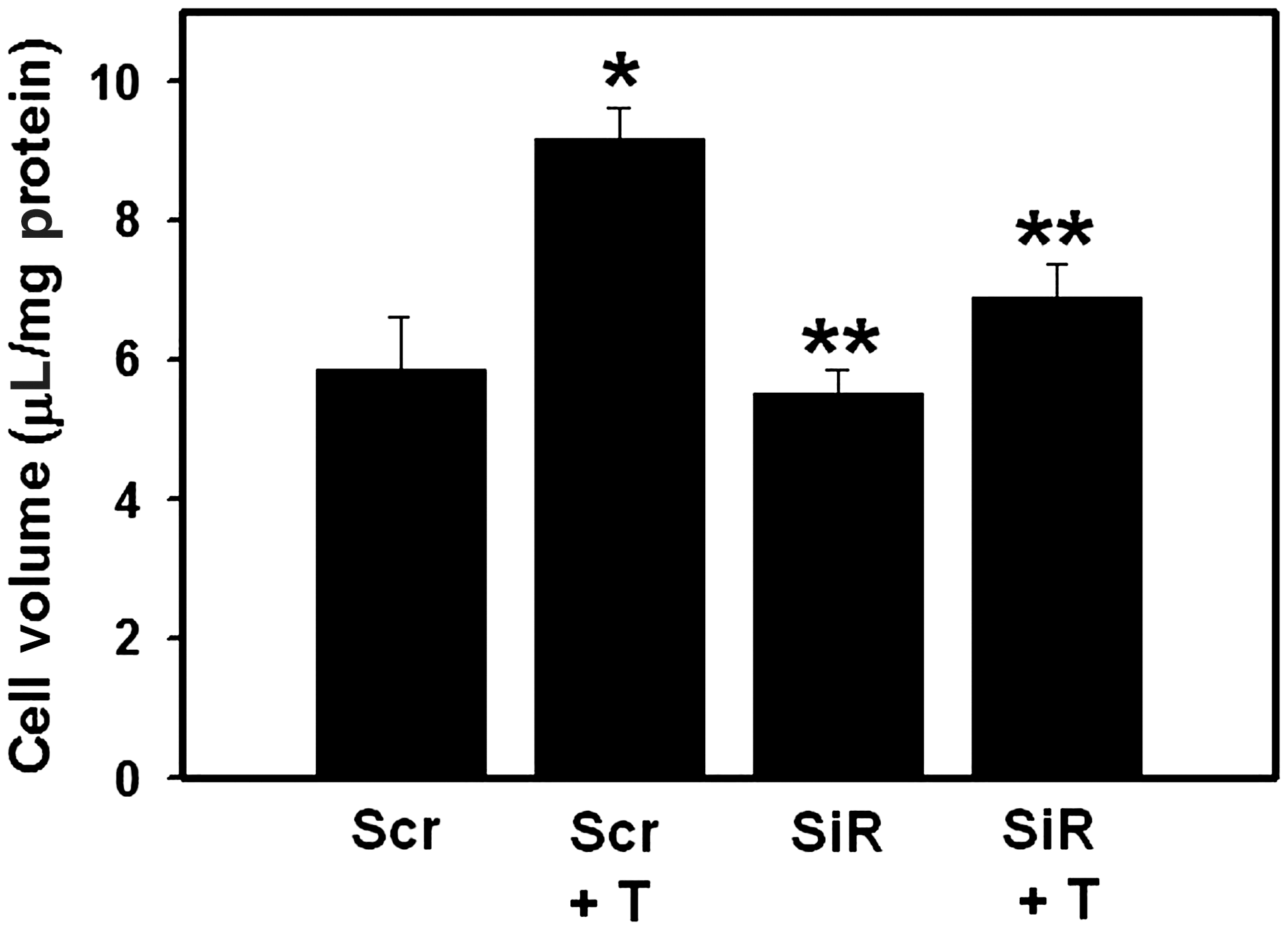
Effect of aquaporin-4 (AQP4) gene silencing on trauma-induced astrocyte swelling. Cultures were transfected with scrambled siRNA (Scr) and AQP4 siRNA (SiR) for 48 h, and cell volume was determined 3 h after trauma (T). Values are mean ± standard error of the mean of five individual plates taken from two separate seedings (*p < 0.01 versus; **p < 0.05 versus Scr + T Scr).
Antioxidants and nitric oxide synthase inhibition diminish trauma-induced AQP4 upregulation
We earlier documented that trauma results in oxidative/nitrative stress, and that antioxidants as well as the nitric oxide synthase inhibitor L-NAME significantly inhibited trauma-induced astrocyte swelling (Jayakumar et al., 2008). We therefore examined the effect of antioxidants and L-NAME on AQP4 protein expression. Cultures were traumatized and immediately thereafter, cells were treated with N-tert-butyl-α-phenylnitrone (PBN; 100 μM), vitamin E (150 μM), cell permeant catalase (500 U/mL), and L-NAME (250 μM), and AQP4 protein levels were determined 3 h after trauma. Vitamin E, PBN, catalase, and L-NAME significantly (40–60%) blocked AQP4 expression (Fig. 5). We previously documented that cultured astrocytes treated with the above antioxidants alone had no effect on AQP4 protein expression (Rama Rao et al., 2010).

Effect of antioxidants on trauma-induced aquaporin-4 (AQP4) protein content in the plasma membrane of astrocyte cultures 3 h after trauma: catalase (CAT, 500 U/mL), vitamin E (Vit E, 150 μM), N-tert-butyl-α-phenylnitrone (PBN; 100 μM), and the nitric oxide synthase inhibitor L-NAME (LN, 250 μM). Values are mean ± standard error of the mean of three individual plates taken from two separate seedings (*p < 0.01 versus control; **p < 0.05 versus trauma; T, trauma).
Inhibition of the mitochondrial permeability transition attenuates trauma-induced AQP4 upregulation
A recent study documented that treatment of cultured astrocytes with cyclosporin A (CsA), an inhibitor of the mPT, significantly blocked trauma-induced astrocyte swelling (Jayakumar et al., 2008). Cultured astrocytes were therefore pretreated with CsA (1 μM) for 10 min. The cells were traumatized and levels of AQP4 were examined 3 h after trauma. CsA completely blocked the upregulation of AQP4 by trauma. To rule the possibility that the effect of CsA might have been mediated by its well-known calcineurin inhibitory effect, cultures were treated 10 min before trauma with FK 506 (5 μM), an inhibitor of calcineurin, and found that such treatment had no effect on AQP4 upregulation by trauma (Fig. 6). These data suggest that CsA's effect in blocking AQP4 upregulation was mediated by inhibition of the mPT.

Effect of cyclosporin A (CsA, 1 μM) on aquaporin-4 (AQP4) protein content in the plasma membranes of astrocyte cultures 3 h after trauma. CsA completely diminished AQP4 upregulation, whereas FK506 (FK, 1 μM), a calcineurin inhibitor that had no effect on the mPT, did not inhibit AQP4 upregulation. Values are mean ± standard error of the mean of three individual plates taken from two separate seedings (*p < 0.01 versus controls; **p < 0.05 versus trauma; T, trauma).
MAPK kinase inhibitors attenuate trauma-induced AQP4 upregulation
We recently showed that trauma results in a significant activation of MAPKs (ERK1/2, JNK1, and p38-MAPK) in cultured astrocytes exposed to FPI, and that inhibition of MAPK activation reduced trauma-induced astrocyte swelling (Jayakumar et al., 2008). We therefore examined the effect of MAPK activation on AQP4 expression. Treatment of cultured astrocytes with UO126 (UO, 10 μM), an inhibitor of ERK1/2, SP 600125 (1 μM), an inhibitor of JNK, and SB 203580 (10 μM), an inhibitor of p38 MAPK and ERK1/2, partially blocked (40–50%) the increase in AQP4 protein expression 3 h after trauma. (Fig. 7). We previously documented that cultured astrocytes treated with the above inhibitors of MAPKs alone had no effect on AQP4 protein expression (Rama Rao et al., 2010).
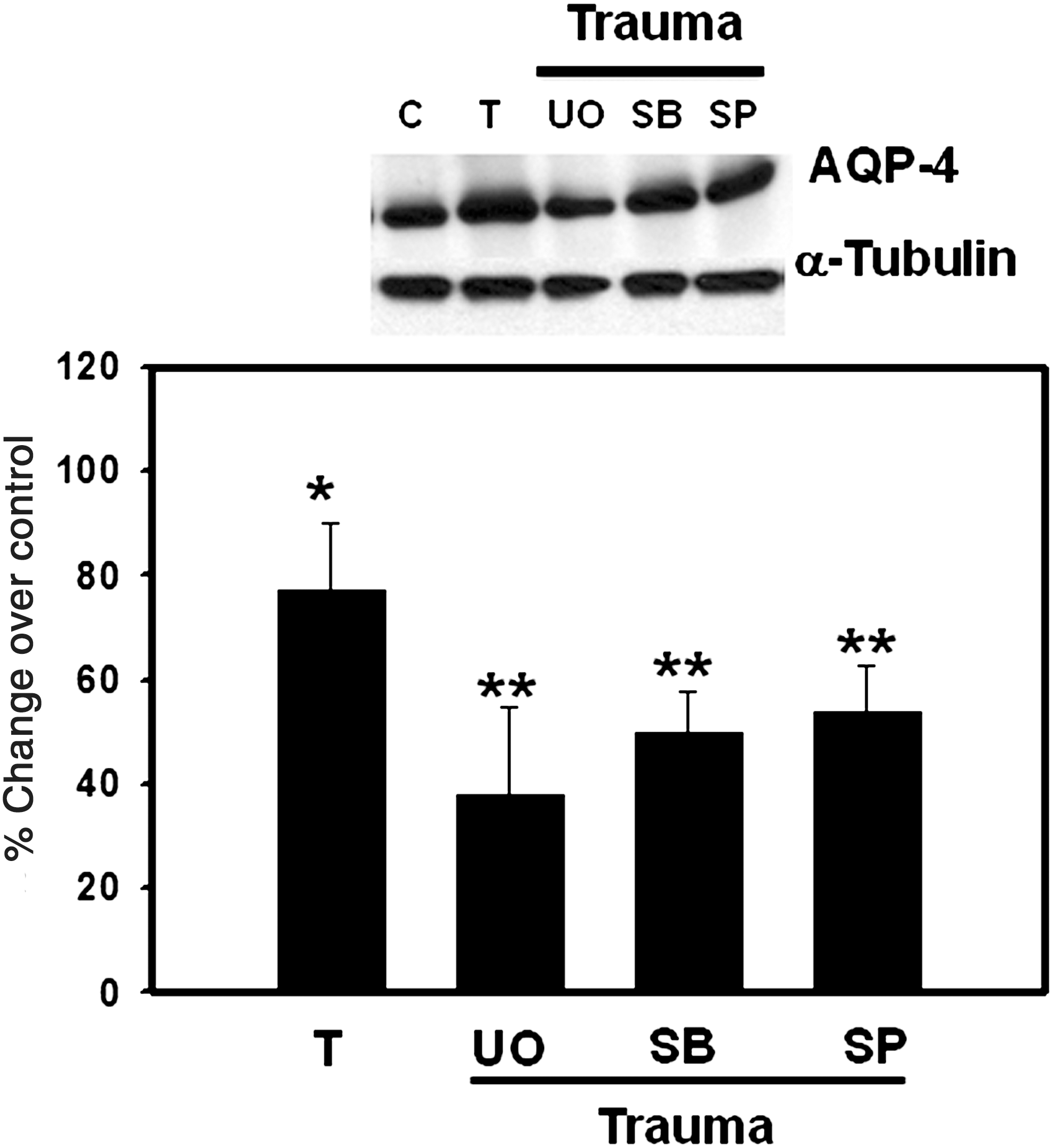
Effect of mitogen-activated protein kinase (MAPK) inhibitors on aquaporin-4 (AQP4) expression in cultured astrocytes 3 h after trauma: UO126 (UO, 10 μM), an inhibitor of ERK1/2, SB239063 (SB, 10 μM), an inhibitor of p38-MAPKs, and SP600125 (SP, μM), an inhibitor of Jun N-terminal kinase (JNK). Values are mean ± standard error of the mean of three individual plates taken from two separate seedings (*p < 0.01 versus control; **p < 0.05 versus trauma; T, trauma).
Inhibition of nuclear factor-kappa B reduces trauma-induced AQP4 upregulation
Several studies have documented that TBI results in the activation of the transcriptional factor nuclear factor-kappa B (NF-κB) (Hang et al., 2006; Jin et al., 2008; Ye et al., 2008). Additionally, NF-κB has been implicated in brain edema and astrocyte swelling in ammonia toxicity (Sinke et al., 2008). Given these findings, we examined the potential role of NF-κB in the AQP4 upregulation by FPI. Pre-treatment (10 min) of cultures with BAY 11-7082 (5 μM), an inhibitor of NF-κB, resulted in a significant (80%, p < 0.01) reduction in trauma-induced AQP4 upregulation (Fig. 8).
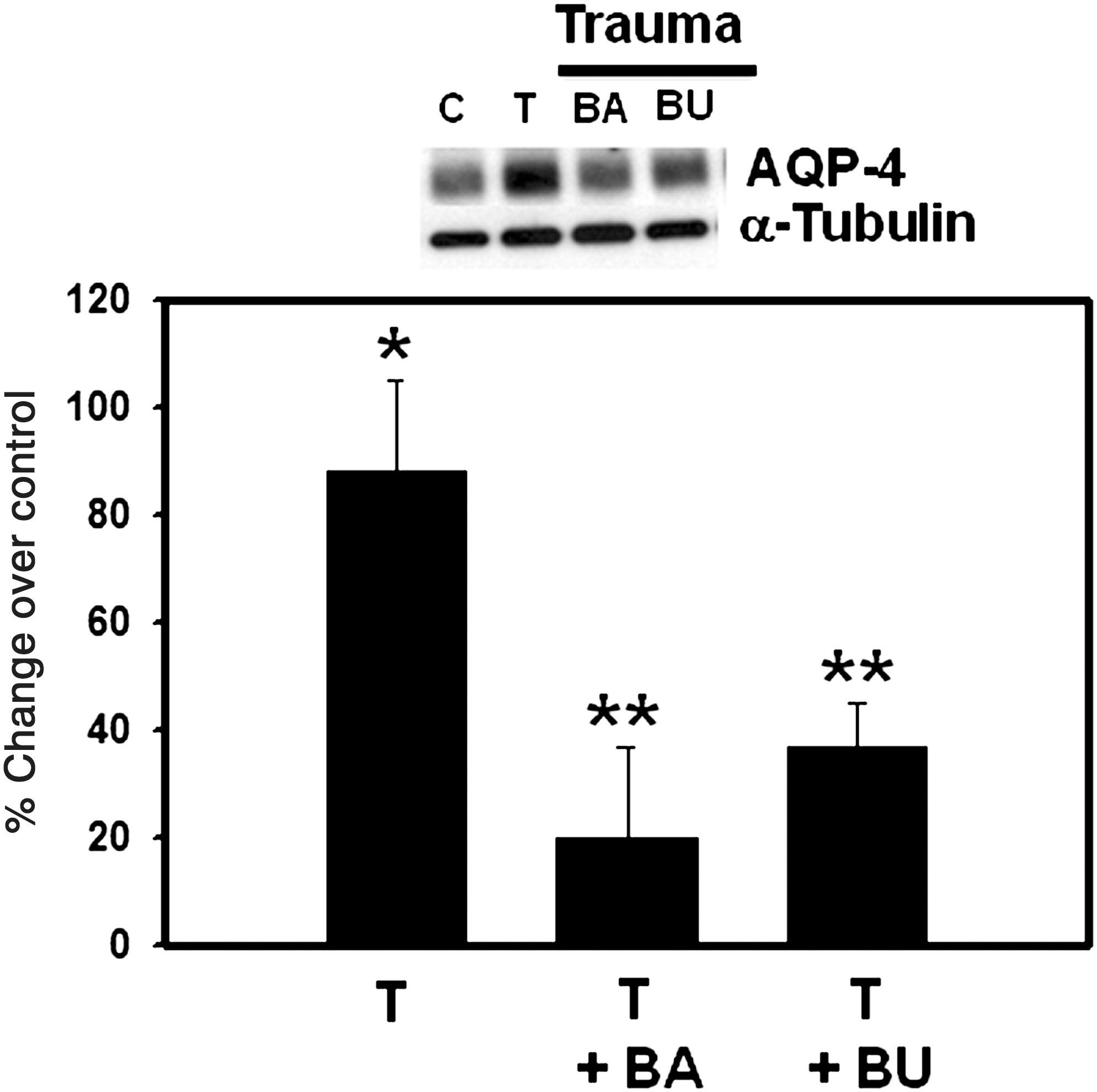
Effect of BAY 11-7082 (BA; 5 μM) and bumetanide (BU; 100 μM) on aquaporin-4 (AQP4) protein in cultured astrocytes 3 h after trauma. Values are mean ± standard error of the mean of three individual plates taken from two separate seedings (*p < 0.01 versus control; **p < 0.05 versus trauma; T, trauma).
Inhibition of the Na+,K+,Cl− co-transporter reduces trauma-induced AQP4 upregulation
TBI has been shown to activate NKCC in rats, and that bumetanide, an inhibitor of NKCC, significantly reduced brain edema in TBI (Lu et al., 2007), suggesting the involvement of NKCC in the brain edema associated with TBI. Consistent with these findings, we recently found that the activity of NKCC in cultured astrocytes was increased following FPI, and that treatment of cultures with bumetanide significantly diminished trauma-induced astrocyte swelling (Jayakumar et al., in press). Since a disturbance in ion homeostasis caused by activation of ion transporters (e.g., NKCC) may lead to the creation of an osmotic gradient resulting in astrocyte swelling, the present study examined the effect of bumetanide on AQP4 upregulation following FPI. Treatment with bumetanide (100 μM) significantly (60%, p < 0.01) inhibited trauma-induced AQP4 upregulation (Fig. 8).
Discussion
This study demonstrates that direct mechanical injury to astrocytes in culture upregulates AQP4 protein in the plasma membrane in a time-dependent manner with a peak at 3 h. Pre-treatment of cultures with cycloheximide resulted in a marked inhibition of AQP4 upregulation, indicating that neosynthesis is a major mechanism responsible for the increase in AQP4 protein expression. Additionally, cultured astrocytes transfected with siRNA targeted to AQP4 showed a significant reduction in trauma-induced astrocyte swelling. Various factors (ONS, MAPKs, mPT, NF-κB, and NKCC), previously reported to be activated by trauma, and which have been shown to contribute to trauma-induced astrocyte swelling, were involved in the upregulation of AQP4.
Brain edema is potentially a lethal complication accounting for >50% of mortality in patients with TBI (Marmarou, 2003). The degree of edema detected in patients on the diffusion-weighted MRI obtained shortly after injury, highly correlates with a negative clinical outcome (Marmarou, 2003). Treatment options for the edema seen following trauma are currently limited, and for the most part ineffective (Marmarou, 2007).
The edema seen in the early phase of TBI is cytotoxic (intracellular fluid accumulation) due to a disturbance in cell volume regulation, while vasogenic edema (extracellular fluid accumulation) due to a breakdown of the blood–brain barrier, represents the major component of the edema in the later phase of TBI (Unterberg et al., 1997). Neuroimaging (MRI) studies support a cytotoxic mechanism for the brain edema, as shown by decreased apparent diffusion coefficient values in the early phase of TBI (Barzo et al., 1997; Marmarou, 2003; Unterberg et al., 2004). The cytotoxic edema in TBI is largely due to swelling of astrocytes (Kimelberg, 1995). Consistent with the above reports, we recently documented that FPI to cultured astrocytes results in significant cell swelling as early as 1 h after trauma (Jayakumar et al., 2008).
Although molecular mechanisms of cytotoxic edema/astrocyte swelling in various neurological conditions remains unclear, aquaporin (AQP) water channels, particularly AQP4, have been strongly implicated in the development of brain edema (for reviews, see King and Agre, 1996; Papadopoulos, et al., 2002). Most of the AQP4 in the brain is present in perivascular astrocyte processes that surround the brain capillaries (Amiry-Moghaddam and Ottersen, 2003; Nielsen et al., 1997). Since astrocytes are critically involved in the maintenance of brain water and ion homeostasis, such strategic localization of AQP4 channels has been suggested to contribute to brain edema formation, possibly explaining the propensity of astrocytes to undergo swelling following various CNS injuries. Thus, mice deficient in brain AQP4 display resistance to the development of brain edema in ischemic stroke and water intoxication (Manley et al., 2000). Conversely, mice overexpressing AQP4 in astrocytes show an accelerated development of brain edema under hypo-osmotic conditions (Yang et al., 2008).
Changes in AQP4 protein and mRNA expression have been reported in various experimental models of TBI. The majority of reports found increased AQP4 expression in FPI (Sun et al., 2003), and after controlled cortical impact (Guo et al., 2006), as well as in closed head injury (Ding et al., 2009; Okuno et al., 2008; Taya et al., 2008, 2010). However, a few reports described a reduction in AQP4 expression following TBI (Ke et al., 2001; Kiening et al., 2002; Zhao et al., 2005). The reason for such a discrepancy relative to changes in AQP4 expression following TBI is not clear. However, variation in the areas examined (epicenter versus penumbra) may partly explain the disparate findings, as Zhao and associates (2005) reported a decrease in AQP4 expression in the core area of the injury, a likely outcome as the tissue was undoubtedly necrotic. On the other hand, an increase was observed in the penumbral region, where the tissue is relatively intact (Ke et al., 2001). Additionally, the nature of the edema present (cytotoxic versus vasogenic) may also influence AQP4 expression levels, as increased AQP4 expression contributes to the development of cytotoxic edema, whereas its reduction is a common feature of vasogenic edema (Ke et al., 2001; Manley et al., 2004).
Our present data showing increased AQP4 expression strongly correlated with our previous observation of the time course of cell swelling in cultured astrocytes following trauma (Jayakumar et al., 2008) (r 2 = 0.9), consistent with a key role of AQP4 in the development of astrocyte swelling. Further support for the role of AQP4 in astrocyte swelling following trauma is the observation in the present study of a marked reduction in trauma-induced astrocyte swelling in cultures transfected with AQP4 siRNA.
Oxidative/nitrative stress
The mechanisms by which trauma results in the upregulation of AQP4 are not known. However, one possible factor is ONS. TBI is known to induce ONS (Cherian et al., 2004; Hall 1993; Raghupathi, 2004). Increased brain nitric oxide (NO) levels and protein tyrosine nitration have also been reported in TBI (Darwish et al., 2007; Unterberg et al., 2004). An increase in free radical production was previously shown in cultured astrocytes exposed to FPI (Panickar et al., 2002). Further, ONS has been implicated in the brain edema/astrocyte swelling associated with various neurological conditions, including TBI (Bauer et al., 1999; Bemeur et al., 2005; Chan et al., 1995; Norenberg et al., 2005).
The present study showing a significant reduction in trauma-induced AQP4 upregulation by antioxidants and the nitric oxide synthase (NOS) inhibitor L-NAME, adds support to the view that ONS is a major factor contributing to the upregulation of AQP4 by trauma. Our findings are also consistent with a previous report showing the upregulation of AQP4 by hydrogen peroxide in cultured astrocytes (Arima et al., 2003). Similarly, aminoguanidine, an inhibitor of inducible nitric oxide synthase, reduced the AQP4 upregulation seen following brain tissue resection (Hao et al., 2009). The antioxidant melatonin has been shown to mitigate AQP4 upregulation in ischemic stroke (Kaur et al., 2006), and curcumin, a compound known to possess antioxidant properties, was shown to inhibit AQP4 upregulation following brain trauma (Laird et al., 2010). We recently found that various antioxidants and L-NAME caused a significant reduction of AQP4 protein levels in the plasma membrane in cultured astrocytes exposed to toxic levels of manganese (Rao et al., 2010). Taken together, these findings strongly suggest that ONS plays a significant role in AQP4 upregulation in various neurological conditions.
Nuclear factor-kappa B
NF-κB is a ubiquitous transcriptional factor activated in response to oxidative stress as well as by inflammatory mediators (Baldwin, 1996). Activation of NF-κB results in induction of several inflammatory/oxidative stress responsive genes, leading to an exacerbation of ONS (Baldwin, 1996). Activation of NF-κB has been documented in TBI (Hang et al., 2006; Jin et al., 2008; Ye et al., 2008). Consistent with these findings, we recently observed that FPI to cultured astrocytes resulted in the activation of NF-κB, while inhibition of such activation significantly blocked trauma-induced astrocyte swelling (Jayakumar et al., in press). The involvement of NF-κB in cell swelling has also been documented in cultures exposed to the neurotoxin ammonia, the principal agent implicated in hepatic encephalopathy (Sinke et al., 2008), and in the formation of brain edema in acute liver failure (Jayakumar et al., in press).
This study found that BAY 11-7082, an inhibitor of NF-κB, decreased AQP4 upregulation. These findings are consistent with a previous study demonstrating that the inflammatory cytokine IL-1β upregulates AQP4 mRNA and protein expression in cultured astrocytes by a mechanism involving NF-κB (Ito et al., 2006). Notably, IL-1β has been shown to result in ONS (Sharma et al., 2007). It is therefore likely that the influence of NF-κB in AQP4 upregulation by trauma involves ONS.
Mitogen-activated protein kinases
MAPKs are stress-responsive signaling kinases commonly activated by ONS (Gaitanaki et al., 2003). MAPKs phosphorylate enzymes and proteins involved in free radical production and associated ONS (Nozaki et al., 2001). Several studies have described the activation of MAPKs as early as 2 h following TBI (Raghupathi et al., 2003). Activation of these kinases has also been reported in cultured astrocytes in a stretch model of injury (Mandell et al., 2001; Neary et al., 2003), as well as following FPI (Jayakumar et al., 2006). It is of interest that inhibitors of all three MAPKs significantly blocked cell swelling in cultured astrocytes following FPI (Jayakumar et al., 2008). The present study showed a significant reduction in trauma-induced AQP4 upregulation by inhibitors of MAPKs, and is consistent with the involvement of MAPKs in the upregulation of AQP4.
The mitochondrial permeability transition
The mPT is a phenomenon that occurs as a consequence of the opening of the permeability transition pore in the inner mitochondrial membrane, a process generally triggered by the mitochondrial overload of Ca2+ (Haworth and Hunter, 1979; Hunter et al., 1976). The mPT is inhibited by cyclosporin A (Broekemeier et al., 1989). The involvement of the mPT in TBI has been demonstrated by the CsA-sensitive swelling of brain mitochondria (Scheff and Sullivan, 1999). Additionally, CsA was shown to reduce the extent of tissue injury after TBI (Scheff and Sullivan, 1999; Sullivan et al., 1999), and to attenuate trauma-induced cell swelling in cultured astrocytes (Jayakumar et al., 2008).
Our findings showing a complete blockade of AQP4 upregulation by CsA, but not by FK 506, in cultures exposed to FPI suggest that the mPT may be involved in AQP4 upregulation. While the mechanism for this observation is not known, it is likely that ONS may also be involved, as the mPT results in increased reactive oxygen and nitrogen species production (Votyakova and Reynolds, 2005; Zorov et al., 2000, 2006).
Na+,K+,Cl− co-transporter
The NKCC is an electroneutral ion transporter involved in the inward transport of Na+/K+ coupled to Cl− transport. The NKCC is mainly involved in cell volume regulation, particularly in states of dehydration and cell shrinkage (Russell, 2000). A recent study documented increased NKCC activity in the choroid plexus of rats following TBI, and that treatment with bumetanide, an inhibitor of NKCC, significantly reduced the brain edema in TBI (Lu et al., 2007).
In the present study, treatment of cultures with bumetanide resulted in a significant inhibition of AQP4 upregulation following FPI, suggesting that NKCC activation influences the AQP4 upregulation by trauma. The mechanism by which NKCC activation causes the upregulation of AQP4 is not known, but it has been speculated that a disturbance in osmotic gradients created by increased activation of ion transporters may trigger a signaling event culminating in the upregulation of AQP4 (Pasantes-Morales and Cruz-Rangel, 2009). Nevertheless, future studies are required to precisely delineate the mechanism by which NKCC contributes to the upregulation of AQP4.
While ONS may represent a central event in the upregulation of AQP4 by the various factors examined in this study (MAPKs, NF-κB, and mPT), we cannot rule out that such factors may impact on AQP4 expression by mechanisms independent of ONS. For instance, it has been observed that exon 1 in the promoter region of aqp4 contained a putative NK-κB cis-acting binding site, suggesting that NF-κB may act on the AQP4 promoter, leading to its transcriptional upregulation (Ito et al., 2006). Likewise, induction of the mPT can result in bioenergetic failure, leading to altered activities of various ion transporters and exchangers that may create an osmotic gradient that triggers the upregulation of AQP4.
In summary, our findings indicate that exposure of astrocytes to FPI results in a significant upregulation of AQP4 protein in the plasma membrane, and that pre-treatment of cultures with cycloheximide blocks the trauma-induced AQP4 upregulation, indicating that neosynthesis is principally responsible for the upregulation of AQP4. FPI to cultured astrocytes transfected with siRNA targeted to AQP4 resulted in a significant reduction in trauma-induced astrocyte swelling, strongly implicating AQP4 in the mechanism of trauma-induced astrocyte swelling. Various factors (ONS, NF-κB, MAPKs, and the mPT), that are known to be activated by trauma and to contribute to trauma-induced astrocyte swelling, also significantly enhanced the upregulation of AQP4. Further, NKCC, which is activated in TBI, was also shown to contribute to the upregulation of AQP4. Targeting AQP4 and factors that regulate it may represent a useful strategy to mitigate the brain edema/astrocyte swelling associated with TBI.
Footnotes
Acknowledgments
This work was supported by a Merit Review from the Department of Veterans Affairs. We are grateful to Alina Fernandez for preparation of astrocyte cultures.
Author Disclosure Statement
No competing financial interests exist.