
Abstract
Although chronic sleepiness is common after head trauma, the cause remains unclear. Transcranial magnetic stimulation (TMS) represents a useful complementary approach in the study of sleep pathophysiology. We aimed to determine in this study whether post-traumatic sleep-wake disturbances (SWD) are associated with changes in excitability of the cerebral cortex. TMS was performed 3 months after mild to moderate traumatic brain injury (TBI) in 11 patients with subjective excessive daytime sleepiness (EDS; defined by the Epworth Sleepiness Scale ≥10), 12 patients with objective EDS (as defined by mean sleep latency <5 on multiple sleep latency tests), 11 patients with fatigue (defined by daytime tiredness without signs of subjective or objective EDS), 10 patients with post-traumatic hypersomnia “sensu strictu,” and 14 control subjects. Measures of cortical excitability included central motor conduction time, resting motor threshold (RMT), short-latency intracortical inhibition (SICI), and intracortical facilitation to paired-TMS. RMT was higher and SICI was more pronounced in the patients with objective EDS than in the control subjects. In the other patients all TMS parameters did not differ significantly from the controls. Similarly to that reported in patients with narcolepsy, the cortical hypoexcitability may reflect the deficiency of the excitatory hypocretin/orexin-neurotransmitter system. These observations may provide new insights into the causes of chronic sleepiness in patients with TBI. A better understanding of the pathophysiology of post-traumatic SWD may also lead to better therapeutic strategies in these patients.
Introduction
S
In the present study, we applied TMS in patients affected by different SWD after TBI in an attempt to investigate whether changes in cortical excitability are associated with the development of post-traumatic EDS.
The following TMS parameters were evaluated: (1) the resting motor threshold (RMT), which reflects the excitability of a central core of neurons and depends on membrane excitability (Hallett, 2000; Ziemann et al., 1996); (2) the central motor conduction time, which reflects the integrity of the corticospinal tract (Rossini et al., 1994); (3) the intracortical inhibition to paired TMS, which is believed to reflect the excitability of inhibitory gabaergic cortical circuits (Di Lazzaro et al., 2000; Ziemann et al., 1996); and (4) the intracortical facilitation to paired TMS, which is thought to depend upon the activity of intracortical glutamatergic excitatory circuits and is also modulated by gabaergic inputs (Kujirai et al., 1993; Ziemann et al., 1998).
Methods
Patients
Severity of TBI was assessed clinically using the Glasgow Coma Scale (GCS; 15=minor, 13–14=mild, 9–12=moderate, and 3–8=severe), and by cerebral computed tomography using the Marshall criteria (I=no visible intracranial pathology, II–IV=midline shift, and V=mass lesion; Marshall et al., 1992). The patients were also given a comprehensive neurological examination.
Only patients with no visible intracranial pathology on cerebral computed tomography (Marshall I) and without pathological findings in the neurological examination were included in the study.
SWD were identified by means of interviews, questionnaires, and sleep studies. During the interviews we assessed changes in sleep habits, sleep quality, and daytime vigilance. Specific SWD were diagnosed according to the international criteria (American Academy of Sleep Medicine, 2005). All patients filled out an extensive sleep questionnaire that comprised validated scales such as the Epworth Sleepiness Scale (ESS; ≥10 suggests EDS; Johns, 1991), the Sleep Apnea Scale of the Sleep Disorders Questionnaire (SA-SDQ; ≥32 for females or ≥36 for males suggests sleep-related breathing disorders; Douglass et al., 1994), and the Ullanlinna Narcolepsy Scale (≥14 suggests narcolepsy Hublin et al., 1994).
Conventional overnight polysomnography (using a 16-channel recording system: Embla A10) was performed from 11
Neuropsychological functions were assessed using the Folstein Mini-Mental State Examination (MMSE; Folstein and Folstein, 1975).
To detect depression, all patients were given the Beck Depression Inventory (BDI; Beck et al., 1961), a self reporting rating inventory measuring characteristic symptoms and severity of depression.
Sixty-seven subjects were diagnosed with post-traumatic SWD and screened for possible study inclusion. Patients (n=3) suffering from post-traumatic epilepsy, as well as patients (n=8) with sleep-wake and/or psychiatric disorders diagnosed prior to TBI, in particular with post-traumatic stress disorder or qualifying for depression (BDI score>9), and patients (n=5) with potential causes of SWD other than TBI (such as obstructive sleep apnea, restless legs syndrome, periodic limb movements at sleep, or behaviorally-induced insufficiency sleep syndrome) were excluded from the study. Patients were also excluded (n=7) on the grounds of a concomitant cervical or upper limb injury that could affect peripheral conduction among peripheral nerves and spinal cord, electrophysiological evidence of subclinical radiculopathy or neuropathy, or an epidural or subdural hematoma or a skull fracture, as they could decrease the capacity of TMS to activate the cortex.
A total of 44 patients (M:F 25:19, mean age 43.8 years, range 18–72 years; 42 right-handed) fulfilled the criteria for inclusion in the study: 11 patients (M:F 6:5) with subjective EDS (defined by an ESS score≥10), 12 patients (M:F 7:5) with objective EDS (defined by mean sleep latency<5 on MSLT), 11 patients (M:F 7:4) with fatigue (defined by daytime tiredness reported on interview, without signs of subjective or objective EDS), and 10 patients (M:F 5:5) with post-traumatic hypersomnia “sensu strictu” (increased sleep need of>2 h per 24 h compared to pre-TBI). Twenty-one patients sustained a mild head injury (GCS score 13 or 14), and 23 a moderate head injury (GCS score between 9 and 12).
The main demographic and clinical characteristics of the patients are reported in Table 1.
ESS, Epworth Sleepiness Scale MSLT, multiple sleep latency test; GCS, Glasgow Coma Scale; EDS, excessive daytime sleepiness.
The selected patients did not report hypnagogic hallucinations, cataplexy-like episodes, or sleep paralysis. On the MMSE a median score of 29.2 points was achieved (range 26–30, <27 in 1 patient). The median Ullanlinna Narcolepsy Scale score was 4.4 (range 0–13). The median SA-SDQ score was 22.0 (range 13–32).
The control group consisted of 14 age-matched healthy subjects (M:F 8:6, mean age 44.8 years, range 20–70 years; 13 right-handed).
Administration of all drugs that affect motor cortex excitability was discontinued in patients and controls at least 2 weeks before the study.
Patients and control subjects provided informed consent before participation in the study, which was performed according to the Declaration of Helsinki and approved by the Ethics Committee of the Christian Doppler Clinic.
Transcranial magnetic stimulation
Transcranial magnetic stimulation was performed 3 months after the TBI during the awake state using two high-power Magstim 200 magnetic stimulators (Magstim Co., Whitland, Dyfed, U.K.) connected to a Bistim module throughout all measurements.
A figure-of-eight coil (external loop diameter 90 mm) was held over the dominant motor cortex at the optimum scalp position to elicit motor responses in the contralateral first dorsal interosseous (FDI) muscle. The induced current flowed in a postero-anterior direction. Surface muscle responses were obtained via two 9-mm diameter Ag-AgCl electrodes with the active electrode applied over the motor point of the muscle and the reference on the metacarpophalangeal joint of the index finger. Muscle responses were amplified and filtered (bandwidth 3–3000 Hz) by a D150 amplifier (Digitimer, Welwyn Garden City, Herfordshire, U.K.).
RMT was defined as the minimum stimulus intensity that produced a liminal motor-evoked response (about 50 μV in 50% of 10 trials) at rest. Central motor conduction was calculated by subtracting the peripheral conduction time from spinal cord to muscles from the latency of responses evoked by cortical stimulation with the formula: motor-evoked potential (MEP) latency−(F latency+M latency −1)/2 in msec (Rossini et al., 1994).
Short-latency intracortical inhibition (SICI) and intracortical facilitation (ICF) were studied using the technique of paired magnetic stimulation with a subthreshold conditioning stimulus (Kujirai et al., 1993). Two magnetic stimuli were given through the same stimulating coil, using a Bistim module over the motor cortex, and the effect of the first (conditioning) stimulus on the second (test) stimulus was investigated. The conditioning stimulus was set at an intensity of 90% of RMT. The second, test, shock intensity was adjusted to evoke a motor-evoked potential in the relaxed FDI with an amplitude of approximately 1 mV peak-to-peak. The timing of the conditioning shock was altered in relation to the test shock. Inhibitory interstimulus intervals (ISIs) of 2, 3, and 5 msec, and facilitatory ISIs of 7, 10, and 20 msec were investigated. Ten stimuli were delivered at each ISI. For these recordings, muscle relaxation is very important and the subject was given audiovisual feedback at high gain (50 μV) to assist in maintaining complete relaxation. All trials in which movement artifact occurred were rejected on-line, and that stimulus condition was repeated. The presentation of conditioned and unconditioned trials was randomized.
The amplitude of the conditioned EMG responses was expressed as the percentage of the amplitude of the test EMG responses. The amplitude of the conditioned responses at the three different inhibitory ISIs, and the amplitude of the conditioned responses at the three facilitatory ISIs were averaged separately, obtaining grand mean amplitudes of the inhibitory and of the facilitatory ISIs.
Since cortical excitability is largely influenced by drowsiness or sleep, the patients were studied during full wakefulness, and in a subgroup of patients we checked this by continuously monitoring electroencephalography during the experiment by means of two gold electrodes placed at the Cz and Pz locations.
To clarify a possible spinal or peripheral contribution to the motor cortex excitability parameters, supramaximal stimulation of the ulnar nerve at the wrist was used to assess spinal and peripheral motor excitability. While FDI was relaxed, the peak-to-peak amplitude of F waves (20 trials, the mean values obtained for the single F waves were averaged post-hoc), and compound muscle action potential (maximum of three trials) were determined.
Statistical analysis
The neurophysiological parameters were analyzed separately by means of between-groups one-way analysis of variance (ANOVA). Where significant group effects were present, post-hoc parametric pair-wise comparisons were applied.
Differences in means of BDI score between the different groups were analyzed pair-wise using the Tukey mean separation test.
Spearman rank correlation was used to explore the relationship between TMS measures and different variables (age, GCS, MSLT, and ESS).
A significance threshold of p<0.05 was used for all tests.
Results
The neurophysiological findings are summarized in the Table 2.
Percentage of test response, grand mean of the SICI at the three ISIs studied.
Percentage of test response, grand mean of the ICF at the three ISIs studied.
Bold type indicates abnormal values (significant difference from controls).
M, mean (values); SD, standard deviation; CMCT, central motor conduction time; RMT, resting motor threshold; MSO, maximum stimulator output; SICI, short-latency intracortical inhibition; ICF, intracortical facilitation; CMAP, compound muscle action potential; SWD, sleep-wake disturbance; EDS, excessive daytime sleepiness.
ANOVA revealed a significant group effect for RMT (F(4,53)=3.13, p=0.022) and for SICI (F(4,53)=2.56, p=0.049). For all the other parameters, ANOVAs did not show any significant group effect (Fs(4,53)<1, p>0.86 for all tests). Post-hoc comparisons showed a significantly higher RMT and a significantly increased SICI in patients with objective EDS compared to the control subjects (p=0.001 and p=0.009, respectively; Fig. 1A and B). In the other patients all TMS parameters did not differ significantly from the controls (p>0.05 for all tests).
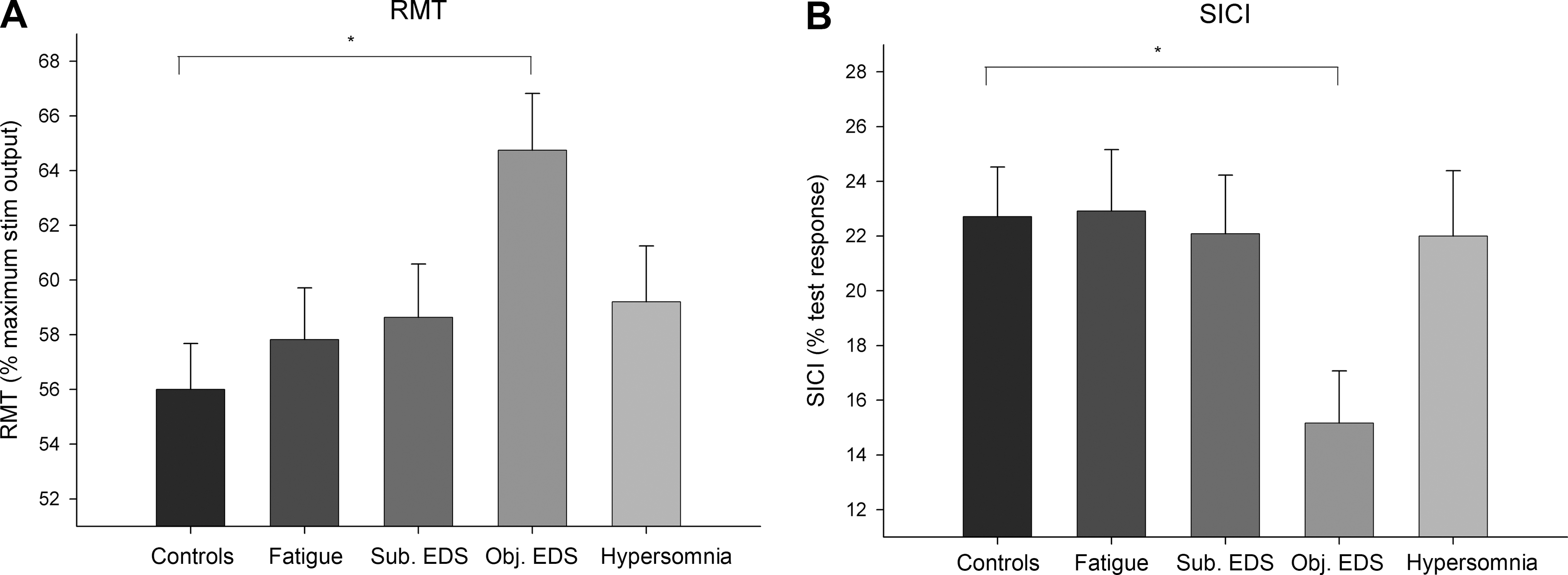
Bar graphs showing mean resting motor threshold (RMT) (
There were no significant differences in the mean BDI scores between the groups (p>0.05).
The Spearman rank correlation analysis revealed a significant correlation between RMT and MSLT (r(42)=−0.46, p=0.002), as well as between SICI and MSLT (r(42)=0.47, p=0.001). No significant correlations between TMS measures and GCS or ESS were found (r<0.25, p>0.11; Fig. 2A and B).
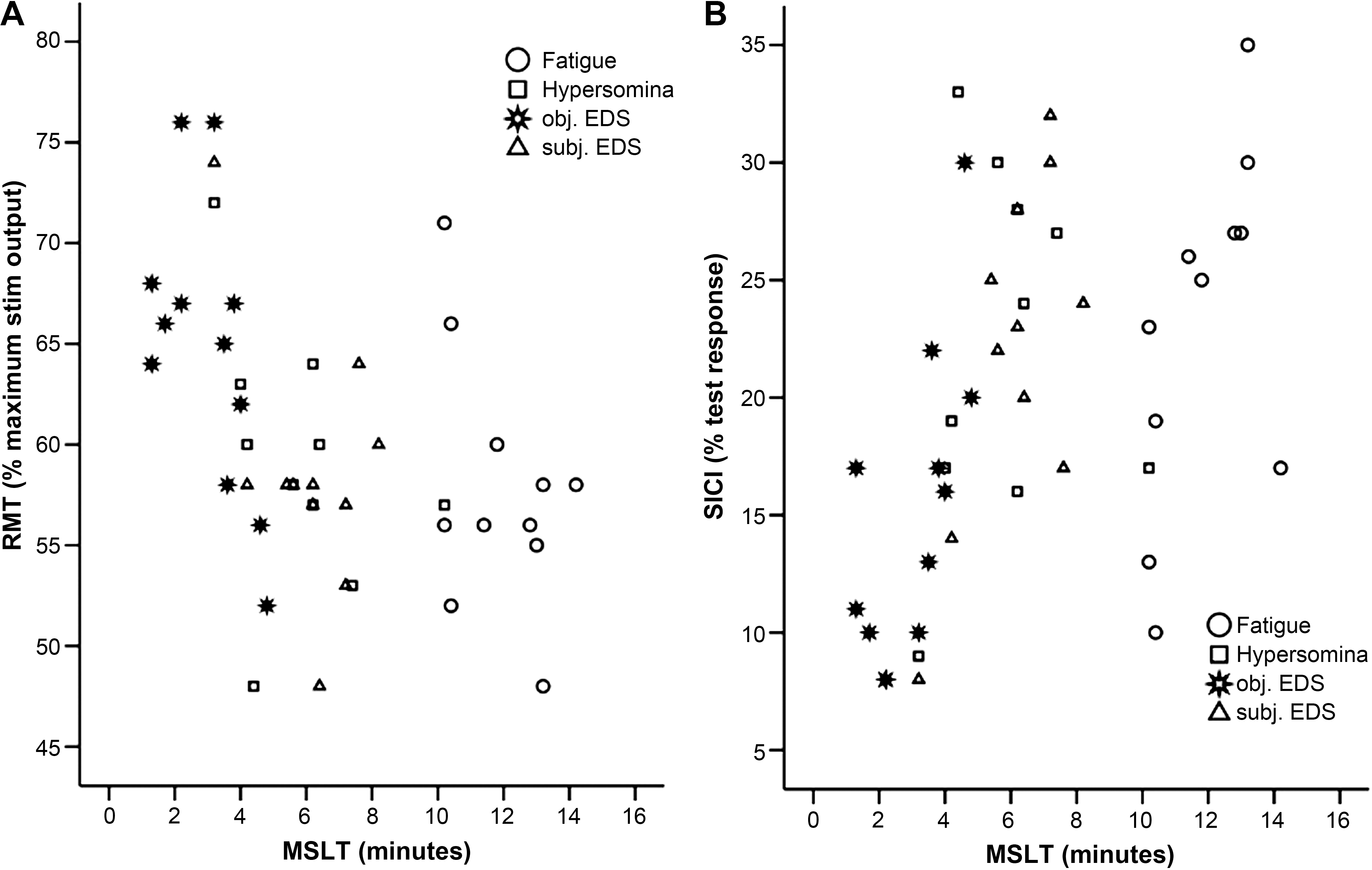
Scatterplots illustrating the correlations between resting motor threshold (RMT) and multiple sleep latency test (MSLT) (
Discussion
The most salient finding of this study was the cortical hypoexcitability (increased RMT and intracortical inhibition to paired TMS) in patients with objective EDS. Both RMT and SICI correlated significantly with the conventional neurophysiological method of MSLT. Interestingly, similar TMS findings have been reported in previous studies on patients with narcolepsy (Oliviero et al., 2005; Nardone et al., 2010; Yoo et al., 2010). Furthermore, Nardone and associates (2010) found in narcoleptic patients a significant correlation between these two TMS measures and the clinically validated measures of daytime sleepiness, the MSLT and the subjective ESS.
A reduced excitability of the corticospinal system during sleep has been explained by the hyperpolarization of the thalamo-cortical neurons deafferenting the cortex from the sensory input at sleep onset (Contreras et al., 1996; Llinas, 1964; Steriade et al., 2001; Timofeev et al., 1996, 2001). The increased RMT in patients with post-traumatic objective EDS can be explained by the occurrence of this hyperpolarization in a dissociated context (eg, during wakefulness, similar to that reported in patients with narcolepsy-cataplexy; Broughton et al., 1986).
Since circuits originating in the lateral hypothalamus widely project to the neocortex, a possible explanation for the increased inhibitory gabaergic activity is the loss of the neuropeptides hypocretin-1 and −2 (also known as orexin-A and -B), resulting in reduced activation of the histaminergic system, which might reduce the tonic inhibition of gabaergic neurons (Huang et al., 2001; Jang et al., 2001; Vincent et al., 1983). It is known that extensive loss of the hypothalamic neurons that produce these neuropeptides causes the severe sleepiness of narcolepsy, and partial loss of these cells may also contribute to the sleepiness of Parkinson's disease and other disorders. Since these hypothalamic neuropeptides are involved in sleep-wake regulation (Nishino et al., 2000), it is reasonable to assume that hypocretin neurotransmitter deficiency could contribute to the pathophysiology of post-traumatic SWD.
Our findings are consistent with recent studies reporting significantly decreased cerebrospinal fluid (CSF) hypocretin-1 levels in patients with acute TBI (Baumann et al., 2005), and a significantly reduced number of hypocretin neurons in patients with severe TBI (Baumann et al., 2009).
Several mechanisms might explain the decrease of CSF hypocretin-1 levels seen after TBI. TBI can injure the hypothalamus, and reduced hypocretin signaling may contribute to the persistent sleepiness often seen in TBI patients. Dysfunctions in the hypothalamus may also occur in a high percentage of patients with mild or moderate TBI (Benvenga, 2000; Crompton, 1971). Therefore, TBI may damage hypocretin neurons or their axons. In patients with increased sleep propensity after TBI, hypocretin production of the remaining neurons may be inadequate, resulting in residual low levels (Gerashchenko et al., 2003; Scammell, 2007a). Since there is evidence that sleep enhances plasticity effects after brain damage (Stickgold et al., 2001), it is reasonable to assume that downregulation of hypocretinergic activity may have beneficial effects on functional outcome.
The abnormal excitability of cortical networks could therefore be the functional correlate of post-traumatic objective EDS and the substrate for allowing dissociated states of wakefulness and sleep to develop suddenly while patients are awake, thus accounting for the narcolepsy-like symptoms. On the contrary, the activity of the wake-promoting neurotransmitter hypocretin does not seem to play a significant pathogenetic role in patients with fatigue and subjective EDS.
It is not clear whether fatigue and EDS belong to the same clinical spectrum; our results suggest that the two symptoms are distinct entities, probably with different pathophysiological pathways.
Surprisingly, also in patients with post-traumatic hypersomnia, we failed to find cortical hypoexcitability. A possible explanation for this finding is that other pathophysiological mechanisms, such as injury to ascending arousal pathways from the brainstem, may contribute to SWD. Indeed, hypersomnolence is common with hypothalamic and rostral brainstem injuries, but is uncommon with selective loss of the hypocretin neurons (Scammell, 2007a). Therefore, it is likely that reduced hypocretin signaling is not the only factor to cause sleepiness after TBI. About one-third of TBI patients have brainstem injuries, often near the midbrain tegmentum, that may disrupt ascending monoaminergic or cholinergic wake-promoting pathways (Crompton, 1971). Loss of the melanin-concentrating hormone neurons may also contribute to the pathophysiology of post-traumatic SWD, as these cells are hypothesized to regulate REM sleep (Adamantidis et al., 2007; Hassani et al., 2009). Overall, it seems likely that the persistent sleepiness in some TBI patients is due to a combination of reduced hypocretin signaling and injury to other sleep-wake regulating systems.
It would be of interest to correlate in future studies the TMS parameters with CSF hypocretin-I levels in patients with post-traumatic SWD.
Bernabeu and associates (2009) recently found that the intracortical inhibition, as revealed by cortical silent period (CSP) duration, did not exhibit any significant differences in patients with traumatic diffuse axonal brain injury. This finding is not necessarily in contrast with ours. Indeed, SICI and CSP are different events mediated by different gabaergic mechanisms. Moreover, the patients of this study had a more severe brain injury and did not present specifically with SWD. On the other hand, significantly more SWD have been reported in patients with mild TBI compared to patients with severe TBI (Mahmood et al., 2004). In agreement with the findings of the prospective study of Baumann and colleagues (2007), in our patients severity of TBI was not associated with the occurrence of the post-traumatic SWD.
Modafinil, a (diphenylmethyl) sulfinyl-2 acetamide derivative, is a new wake-promoting substance used for the treatment of excessive sleepiness, particularly that associated with narcolepsy (Bastuji and Jouvet, 1988; U.S. Modafinil in Narcolepsy Multicenter Study Group, 1998; Scammell, 2007b). The effects of modafinil in patients with sleep disorders after TBI have rarely been investigated (Castriotta et al., 2007); only recently a double-blind, randomized, placebo-controlled study that included electrophysiological laboratory testing showed that 100–200 mg modafinil is effective and well tolerated in the treatment of post-traumatic EDS, but not of fatigue (Kaiser et al., 2010). Interestingly, it has been demonstrated (Nardone et al., 2010; Yoo et al., 2010) that modafinil reverses cortical hypoexcitability in narcoleptic patients. Therefore, stimulants such as modafinil that are helpful in narcolepsy and other hypocretin-deficient disorders should be studied systematically in patients with post-traumatic SWD. TMS might be useful in identifying patients in whom a decrease of cortical excitability occurred, and who would be suitable for treatment with wake-promoting drugs.
In conclusion, we demonstrated that cortical hypoexcitability underlies sleepiness in patients with post-traumatic objective EDS. TMS techniques may help to disclose new pathophysiological aspects of post-traumatic SWD, also providing a rationale for novel treatment options.
Footnotes
Author Disclosure Statement
No conflict of interest exists.