
Abstract
β-Amyloid (Aβ) peptides, most notably associated with Alzheimer's disease, have been implicated in the pathogenesis of secondary injury after traumatic brain injury (TBI). A prior study has demonstrated that blocking the β-site amyloid precursor protein (APP)-cleaving enzyme 1 (Bace1) required for production of Aβ from APP improved functional and histologic outcomes after controlled cortical impact (CCI) in aged mice. However, the majority of patients with severe TBI are young adults under the age of 40. Prior experimental models have suggested age-dependent differences in Aβ clearance, and a recent study in our lab suggests that young animals remediate acute elevations in Aβ after CCI better than older animals. We therefore tested the hypothesis that Bace1 deletion in young adult mice would not be protective after CCI. Male Bace1 knockout (Bace1 −/−) and wild-type Bace1 +/+ (C57BL/6) mice (2–3 months old) were subjected to CCI (n=18–23/group) or sham injury (n=10–12/group). Functional outcomes were assessed with wire grip (motor) and the Morris water maze (MWM; spatial memory). Soluble Aβ levels were assessed at 48 h after CCI. Histopathological outcomes were assessed by lesion and hippocampal volume. Clustered ordinal logistic regression showed overall significant impairment in motor performance in injured Bace1−/− versus Bace1+/+ animals (p=0.003). No significant differences in MWM performance were found on repeated-measures ANOVA (p=0.11) between groups. Probe scores were significantly worse in injured Bace1−/− versus Bace1+/+ mice (p=0.0009). Soluble Aβ40 was significantly lower in ipsilateral hemispheres of Bace1−/− than in Bace1+/+ animals after CCI (0.9 [IQR 0.88–0.94] pmol/g protein versus 3.8 [IQR 2.4–6.0] pmol/g protein; p=0.005). Lesion and hippocampal volumes did not differ between injured groups. The data suggest that therapies targeting Bace1 may need to be tailored according to age and injury severity, as their use may exacerbate functional deficits after TBI in younger or less severely injured patients.
Introduction
Aβ is a 40–42 amino acid peptide implicated in synaptic dysfunction and cell death in Alzheimer's disease (AD; Lue et al., 1999; Venkitaramani et al., 2007). The interest in the role of Aβ after TBI has grown from epidemiological studies that have demonstrated an association between a history of TBI and the development of AD later in life (Graves et al., 1990; Mortimer et al., 1985, 1991; Salib and Hillier, 1997; Szczygielski et al., 2005; Van Den Heuvel et al., 2007). Additionally, post-mortem studies have demonstrated AD-like deposits of Aβ in as many as 30% of TBI victims (Roberts et al., 1991, 1994). These observations have led to interest in Aβ as a therapeutic target after TBI. A recent landmark study by Loane and associates demonstrated that strategies targeting Aβ improved histopathological and functional outcome after TBI in aged mice (Loane et al., 2009).
Despite the promise of this recent investigation, it is unclear whether Aβ is an appropriate therapeutic target across the spectrum of age. Loane and colleagues demonstrated improved outcomes after controlled cortical impact (CCI) in aged mice unable to generate significant Aβ due to blocking of the β- or γ-secretase enzymes required for production of Aβ from amyloid precursor protein (APP; Loane et al., 2009). However, prior investigations have suggested that the effectiveness of at least some therapies may depend on age (McDonald et al., 1988; Osteen et al., 2001), and age-dependent differences in Aβ clearance (Hickman et al., 2008) may modify the efficacy of strategies targeting Aβ-induced secondary injury after TBI. Experimental models have shown that Aβ toxicity increases in both cultured neurons (Brewer, 1998) and microglia (Hickman et al., 2008) with aging. We have recently published a study in a CCl model that suggests that Aβ toxicity after TBI may not be relevant to younger patients after TBI (Mannix et al., 2010), and the majority of severe brain trauma patients are young adults between the ages of 18 and 40 (Langlois et al., 2006). Here we tested the null hypothesis that following CCI, genetic antagonism of Bace1 will not improve functional and histopathological outcome after CCI in young adult mice.
Methods
Mice
All experiments were approved by the Massachusetts General Hospital Institutional Review Board and complied with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Mice were given free access to food and water and were housed in laminar flow racks in a temperature-controlled room with 12-h day/night cycles. Male (2–3 months old) Bace1 knockout (Bace1 −/−) and wild-type Bace1 +/+ (C57BL/6) mice were used as controls (all mice were from Jackson Laboratories, Bar Harbor, Maine). Bace1 is the initial rate-limiting enzyme for Aβ production, and Bace1 −/− mice produce very low levels of Aβ (Cai et al., 2001). Bace1 −/− mice develop normally, are fertile, are grossly phenotypically normal, and are backcrossed into C57BL/6 at least 7 times (Jackson Laboratories, March 1, 2006). In all experiments, male mice 2–3 months of age were used. Postoperatively, brain- and sham-injured animals were housed together by genotype. Experimenters were blinded to genotype and injury status.
Experimental protocols
A total of 32 Bace1−/− and 42 Bace1+/+ mice were used. For all experiments, investigators were blinded to genotype. Bace1−/− and Bace1+/+ mice were genotyped using standard protocols recommended by Jackson Laboratories (stock number 004714).
Mouse controlled cortical impact model
The mouse CCI model was used as previously described with minor modifications to injury severity level outlined below (Mannix et al., 2010). Mice were anesthetized with 3% isoflurane, N2O, and O2 (2:1), and placed in a stereotactic frame. A 5-mm craniotomy was performed over the left parietotemporal cortex and the bone flap was removed. Mice underwent sham injury (craniotomy only, n=10–12/group) or CCI (n=18–23/group) using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/sec, and impact depth of 0.6 mm. The bone flap was discarded, the scalp was sutured closed, and the mice were allowed to recover from anesthesia in their cages. To reduce variability in injury level attributable to environment, all mice were injured by the same experimenter who was blinded to group assignment.
Determination of genotype and brain-soluble Aβ40
Genotype analysis of Bace1 −/− and Bace1 +/+ mice was done immediately after the last Morris water maze (MWM) trial on day 10. In order to confirm the effect of Bace1 deletion, we assessed soluble Aβ40 in Bace1 −/− and Bace1 +/+ 48 h after both CCI (n=4–11/group) and sham injury (n=5–6/group). We assessed Aβ40 levels rather than Aβ42 because Aβ40 levels are normally higher in brain tissue than Aβ42 levels. Soluble Aβ40 was determined in whole ipsilateral hemispheres of sham-injured and injured mice using a sandwich enzyme-linked immunosorbent assay (ELISA; Wako, Richmond, VA) as previously described, using sample protein concentrations of 1–2 mg/mL (Mannix et al., 2010). Data were expressed as pmol/g protein.
Assessment of motor function
Gross vestibulomotor function was assessed using the wire grip test (Mannix et al., 2010) in naïve animals (n=4–6/group) in a single day of testing, and in sham-injured (n=6/group) and CCI (n=17–18/group) animals on days 1–7 after injury. The test consisted of placing the mouse on a wire suspended between two poles and grading the degree of attachment and movement of the mouse. Scores were as follows: 0 was given to a mouse that fell from the wire within 30 sec; 1 point was given for unilateral grasp of either the upper or lower extremities; 2 points were given for midline grasp of both upper and lower extremities but not the tail; 3 points were given for midline grasp of all extremities plus the tail; 4 points were given for movement along the wire after achieving a score of 3; and 5 points were given for a score of 4 plus climbing down the pole within 60 sec.
Spatial memory acquisition assessment
Spatial memory performance was assessed using the MWM as previously described (Mannix et al., 2010; Morris, 1984). MWM trials were conducted on days 7–10 after sham-injury (n=6/group) and CCI (n=17–18/group). A white pool (83 cm diameter and 60 cm deep) was filled with water to a depth of 29 cm. Several highly visible intra- and extra-maze cues that remained constant throughout the trials were located in and around the pool. Water temperature was maintained at approximately 24°C. The goal platform (a round, clear, acrylic glass platform 10 cm in diameter) was positioned 1 cm below the surface of the water. Each mouse was subjected to a maximum of two trials per day. Each trial consisted of four subtrials in which mice started at each of four locations (north, south, east, or west). For each subtrial, mice were placed in the pool in the selected quadrant facing the wall. Mice were given a maximum of 90 sec to find and rest upon the submerged platform. If the mouse did not rest on the platform by 90 sec, it was gently placed there for 10 sec by the experimenter. For probe trials, mice were placed in the pool with the platform removed. The time that the animal swam in the target quadrant was recorded (maximum 60 sec). For visible platform trials, the goal platform was marked by red tape and placed ½ cm above the water level. Performance in the MWM was quantitated by latency to the platform for hidden trials (90 sec maximum), or latency in the target quadrant for probe trials (60 sec maximum). MWM trials were conducted in the following manner: trials 1 and 2 on day 7 after CCI or sham-injury; trials 3 and 4 on day 8 after CCI or sham-injury; trial 5 on day 9 after CCI or sham-injury; probe 1 trial 6 h after trial 5 on day 9 after CCI or sham-injury; and two visible trials and probe trial 2 on day 10 after CCI or sham-injury.
Lesion volume and hippocampal volume loss
Morphometric image analysis was used to determine lesion volume and hippocampal tissue loss after CCI. On day 21 after CCI, mice (n=8/group) were anesthetized with isoflurane, decapitated, and the brains were removed. The brains were fresh frozen and coronal sections (12 μm) were cut every 0.5 mm from the anterior to the posterior brain, mounted on poly-L-lysine-coated slides, and stained with hematoxylin. The area of hemispheres and hippocampi were determined using image analysis (Nikon Eclipse Ti 2000, MS Elements; MVI, Avon, MA). Lesion volume was obtained by subtracting the volume of brain tissue remaining in the left (injured) hemisphere from that of the right (uninjured) hemisphere and expressed in mm3. Hippocampal tissue loss was obtained by the ratio of volume of brain tissue remaining in the left (injured) hippocampus divided by that of the right (uninjured) hippocampus, and reported as percentage of remaining tissue (Loane et al., 2009).
Statistical analyses
Data are mean±standard error of the mean (SEM) or median (interquartile range, IQR). Motor data, which yield ordinal responses, were analyzed with ordinal logistic regression at individual time points, as well as with clustering to account for repeated measures and yield an overall test statistic. MWM data, which are continuous, were analyzed by t-test for individual trials, and by repeated-measures analysis of variance (RM ANOVA) for an overall statistic. Aβ40 levels and volumetric data were analyzed using the rank sum test or ANOVA as appropriate. For all tests, p<0.05 was considered significant except for repeated-measures tests involving multiple comparisons, for which a Bonferroni correction was utilized and p=0.025 (α/n, where α=0.05 and n=2=number of comparisons).
Results
Genotype analysis of Bace1−/− and Bace1 +/+ mice showed the expected bands and confirmed the identity of Bace1−/− and Bace1+/+ mice (data not shown). As previously reported, brain-soluble Aβ40 was significantly decreased in whole hemispheres of Bace1−/− versus Bace1+/+ animals, both after sham injury (0.22 [IQR 0.19–0.53] pmol/g protein versus 1.6 [IQR 1.4–2.9] pmol/g protein; p=0.01), and CCI (0.9 [IQR 0.88–0.94] pmol/g protein versus 3.8 [IQR 2.4–6.0] pmol/g; p=0.005). Bace1 +/+ mice demonstrated increased Aβ40 after CCI compared to sham controls (p=0.03), but the differences in Aβ40 in injured versus sham-injured Bace1−/− mice did not reach statistical difference (p=0.14). There were no differences between Bace1−/− and Bace1+/+ animals in pre-injury weights (p=0.18), or weight loss 1 week after injury (p=0.31).
Motor outcome
There were no baseline differences in motor performance between naïve Bace1−/− versus Bace1+/+ mice (p=0.8). Wire grip scores did not differ overall between CCI or sham-injured Bace1 +/+ animals (p=0.054), but were significantly worse in injured Bace1 −/− compared to injured Bace1 +/+ animals on days 1–4 after injury (p<0.05 at each time point), as well as overall (p=0.003). Injured Bace1 −/− animals also performed worse than sham-injured Bace1 −/− mice on days 1, 3, and 4 after injury or sham injury (p<0.05 for each time point), as well as overall (p<0.001; Fig. 1A).
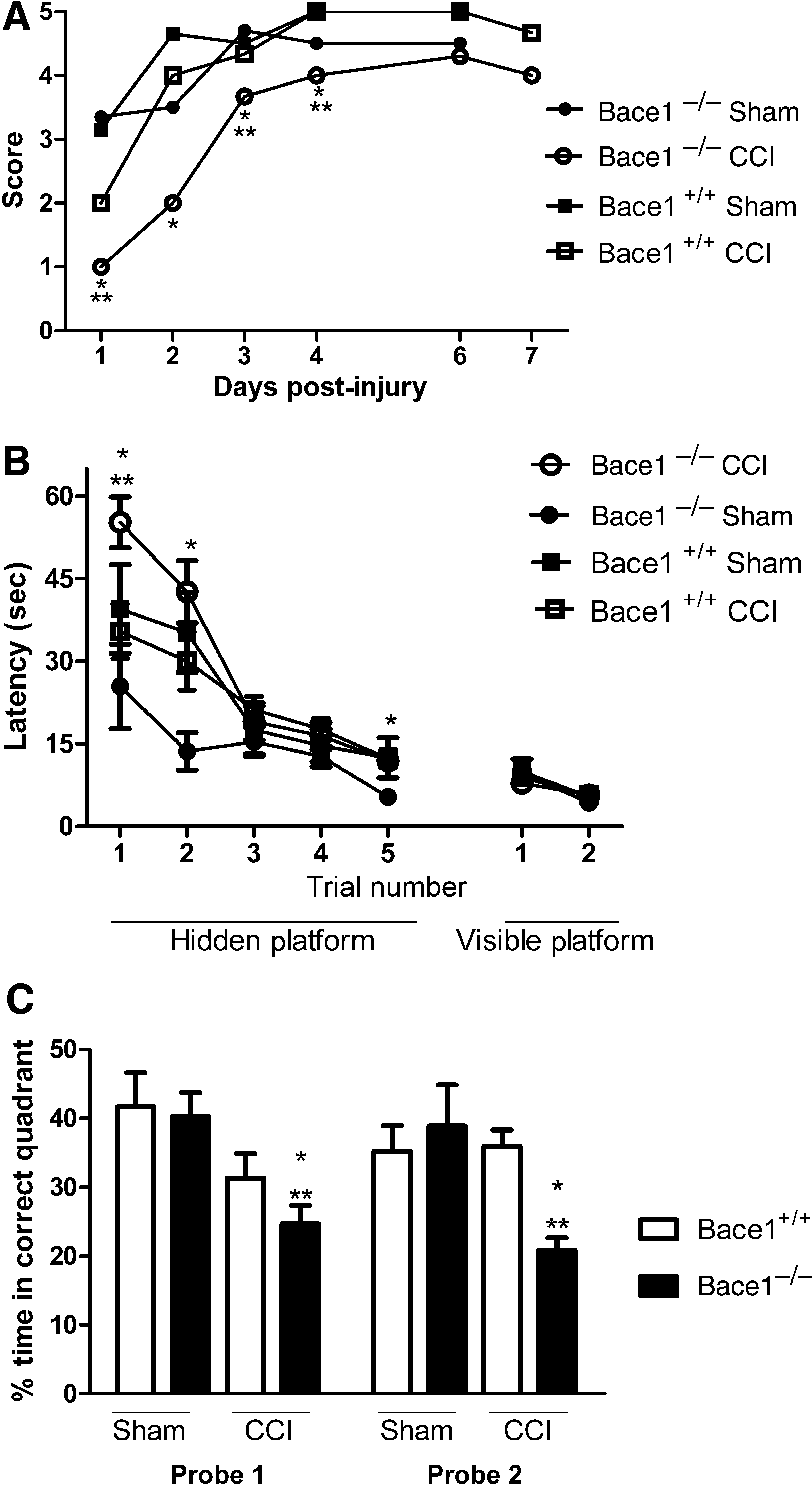
Functional outcomes after controlled cortical impact (CCI) or sham injury. (
Cognitive outcome
All sham-injured and injured groups demonstrated time-dependent improvements in latency to the hidden platform, indicating an ability to learn the MWM paradigm before and after CCI (p<0.001 for time for all groups). Hidden platform trial performance did not differ between sham-injured Bace1 −/− and Bace1 +/+ animals. Hidden platform trial performance also did not differ overall between injured Bace1 −/− and Bace1 +/+ mice (p=0.11), despite significant differences on trial 1 (p=0.007). However, while injured Bace1+/+ animals showed no cognitive deficits compared to their sham controls, injured Bace1−/− mice performed significantly worse than sham-injured Bace1−/− mice on trial 1, trial 2, and trial 5 (p<0.05 for each trial), as well as overall (p=0.0029; Fig. 1B). No differences in visible platform trials were observed among any of the sham-injured and injured groups (Fig. 1B).
Mean probe trial latencies (Fig. 1C) in sham-injured Bace1−/− and Bace1 +/+ animals did not differ between groups. On RM ANOVA, overall probe trial performance was significantly worse in injured Bace1−/− than in Bace1 +/+ mice (p=0.0009). Injured Bace1−/− mice also performed significantly worse than sham-injured Bace1−/− animals (p=0.0005), but injured Bace1 +/+ animals demonstrated no statistically significant difference in performance compared to sham-inured Bace1 +/+ animals (p=0.20).
Histopathology
At 23 days after CCI, Bace1 −/− and Bace1 +/+ mice showed well-demarcated cavitary lesions involving the cortex and hippocampus in injured hemispheres (Fig. 2A). There were no statistically significant differences between the percentage of hippocampal loss (Fig. 2B) or overall lesion volume between groups (Fig. 2C).
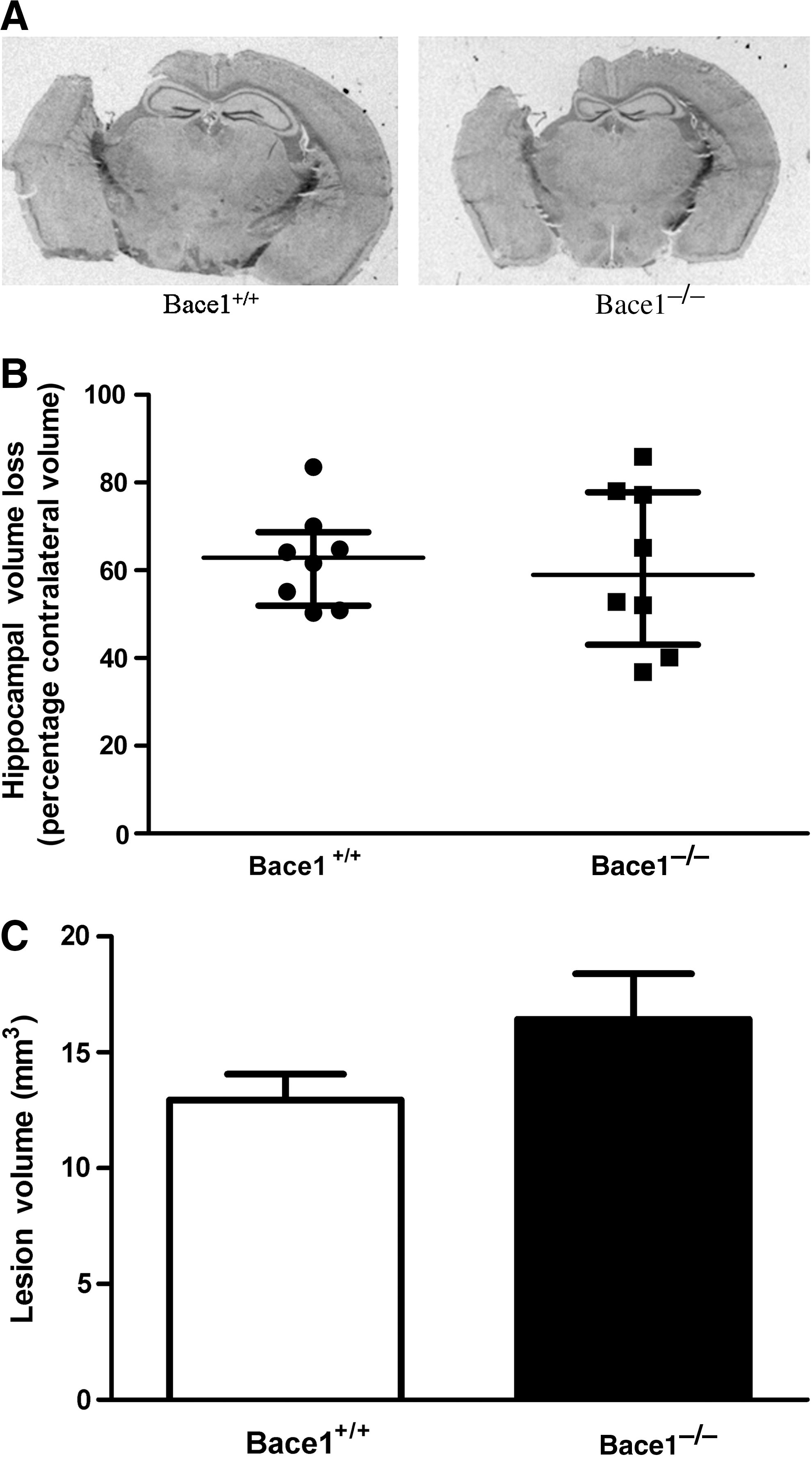
Histological outcomes after controlled cortical impact (CCI). (
Discussion
We report for the first time detrimental effects of Bace1 gene deletion after experimental TBI in young adult mice. The current study was undertaken using a CCI injury level that resulted in no detectable motor or cognitive deficits in injured versus sham-injured wild-type mice. However, significant motor and cognitive deficits were noted in injured Bace1−/− mice compared to both injured Bace1+/+ animals and sham-injured Bace1−/− mice. The detrimental effects of Bace1 deletion on functional performance after CCI was not explained by differences in histopathology or baseline differences in functional performance in Bace1−/− and Bace1+/+ mice.
Our findings are in contrast to a recent report by Loane and associates (2009), who found that Bace1 gene deletion decreased brain tissue damage and improved motor and hidden platform MWM deficits after CCI in aged mice (11–12 months; Loane et al., 2009). The detrimental effects on functional performance and lack of overt tissue protection in Bace1−/− mice in our study is in contrast to the remarkable protection observed in aged mice in the Loane study, despite the comparable levels of overall hemispheric tissue loss and hippocampal damage in Bace1 +/+ mice reported in the two studies.
There are several possible explanations for the divergent results of the current study and those of Loane and colleagues (2009). First and foremost, we used young adult mice in our study compared to the aged mice used in the Loane study. Outcome after TBI is known to be worse in aged adults and harder to effectively treat, due in part to comorbidities associated with aging and normal deterioration of brain function with aging (Frankel et al., 2006; Pennings et al., 1993; Tokutomi et al., 2008). However, we believe using a model with young adult mice is more clinically relevant since young adults comprise the majority of brain-injured patients. Effective treatment could produce lifelong benefits that translate into over 48 billion dollars per year in costs of rehabilitation and lost productivity (Langlois et al., 2006). The current study suggests, however, that Bace1 gene antagonism may not benefit young adult males after TBI. Further experimental studies are clearly needed.
Another explanation for the above-mentioned results is that, in contrast to the Loane study, we used only male mice in our study. Prior studies have demonstrated significant neuroprotective effects of female gender in outcomes after ischemic stroke (Hall et al., 1991). However, other experimental models suggest that female gender increases AD pathology through mechanisms including increased γ-secretase activity with aging (Placanica et al., 2009). It is unclear how Bace1 deletion might alter the interaction of age and gender after TBI, but this is an important clinical question that needs to be addressed in future studies. An additional significant difference between our study and that of Loane and associates (2009) is the level of motor and cognitive deficits seen despite similar lesion size across the two CCI models. We surprisingly did not find an effect of injury on motor performance in Bace1+/+ mice. Differences in MWM performance across labs (despite similar lesion size in CCI) may arise because of differences in environment or testing apparatus that are difficult to control. While our data suggest that Bace1 gene deletion is detrimental after CCI in young adult mice, it is also possible that Bace1 gene deletion may have effects that are injury-level specific. Prior studies in severe TBI patients demonstrate that the type of injury may influence Aβ dynamics (Marklund et al., 2009), and the severity of injury may impact the resultant risk of developing AD (Guo et al., 2000), suggesting that Bace1 gene deletion could have differential effects across the spectrum of injury.
Another important difference between the current study and that of Loane and colleagues is the inclusion of probe trials in the current study. Probe trials are the gold standard for assessment of spatial memory (Gallagher et al., 1993). Loane and associates (2009) did not report probe trial data from injured Bace1−/− and +/+ mice, leaving open the possibility that the beneficial effects that they found in injured aged Bace1−/− mice may be attributable to non-spatial aspects of learning. Finally, it is important to note that the timing of the behavioral testing in our study versus that of Loane and associates may preclude direct comparison of the studies. Although we found a significant detrimental effect of Bace1 deletion on wire grip on days 1–7 after CCI, Loane and colleagues reported a significant beneficial effect of Bace1 deletion on beam walk testing only at days 7–21 after injury. In addition, the Loane group performed MWM testing approximately 1 week later than we did (days 7–10 in our study versus days 15–18 in the Loane study), noting a beneficial effect of Bace1 deletion only on day 18.
The significant differences in results seen between the current investigation and that of the Loane group suggest that more studies are needed to fully characterize the possible age-dependent roles of Bace1 in recovery after TBI. Prior studies have demonstrated that Bace1 activity increases with aging (Miners et al., 2010), and after TBI (Blasko et al., 2004), though it remains unknown how age at injury alters the long-term effects of Bace1 activity and Aβ accumulation after TBI. Advanced age and TBI may synergistically increase Bace1 activity and Aβ accumulation. In addition, older subjects may be more sensitive to the acute effects of increased Bace1 activity and Aβ production after TBI compared to younger subjects. Alternatively, young adults may be more sensitive to the detrimental effects of decreased Bace1 activity after TBI, perhaps due to loss of activity on substrates other than APP (Vassar et al., 2009). It is also possible that Bace1 may confer both protective and detrimental effects after TBI, and that balance is altered with aging. Beneficial effects of Bace1 include enhanced neuronal viability in vitro (Plant et al., 2003), perhaps through the formation of Aβ monomers which have been shown to enhance survival of developing neurons under conditions of trophic deprivation, and to protect mature neurons against excitotoxic death (Giuffrida et al., 2009).
Although we found a detrimental effect of Bace1 deletion on functional outcome after CCI, we did not find histopathological differences between injured Bace1−/− and Bace1 +/+ mice. The lack of correspondence between histopathology and behavioral differences in our study is not unexpected, given the known lack of correlation between histopathology and functional outcome in experimental TBI models (Clark et al., 2000). Somewhat surprisingly, the level of injury that produced a cavitary lesion and some hippocampal damage failed to produce a motor difference in sham-injured and CCI wild-type mice. This is most likely due to some cortical injury that occurred in sham-injured animals during craniotomy that reduced motor baseline function compared to naïve (uninjured) C57 black mice. With respect to cognitive outcome, two explanations are possible. First, our lesion, though still significant, may still be below the level needed to produce cognitive deficits. Second, the use of a 10-cm platform and the specifics of how we performed MWM testing in our lab may not be sensitive enough to detect subtle cognitive deficits at the current level of injury. However, our finding that Bace1−/− mice were not protected from histopathological damage corroborates our lack of evidence of a protective role in functional outcome, as reported for aged Bace1−/− mice (Loane et al., 2009), and suggests that age-dependent effects of Bace1 deletion may not be limited to behavioral differences after TBI.
There are several important limitations and caveats to our study. First, although Bace1−/− mice are inbred on a C56BL/6 background, genetic drift may occur as a result of prolonged inbreeding. This may have contributed to minor differences in genetic background that may have influenced cognitive outcome in Bace1−/− mice after TBI. Second, we did not evaluate other indices of histopathology such as axonal damage and inflammation, which may have contributed to worse functional outcome in Bace1−/− mice. Third, due to technical limitations, we did not evaluate Aβ42, which has been demonstrated to be the predominant and likely toxic species seen after human TBI (DeKosky et al., 2007; Gentleman et al., 1997). Fourth, we demonstrated low levels of Aβ40 in both sham-injured and CCI Bace1 −/− animals. Although Loane and associates do not specify when Aβ40 levels were obtained, they also demonstrated low levels of Aβ40, comparable to the levels seen in sham-injured animals in our study (0.1±0.09 fmol per mg protein in their study versus 0.22 [IQR 0.19–0.53] pmol/g protein in our study). Fifth, by measuring whole ipsilateral brain Aβ40, we did not distinguish between hippocampal and cortical Aβ40 levels, which may offer important insights into the motor and cognitive differences observed in our study, particularly since low levels of Aβ40 were detected in Bace1−/− mice and could be region-specific. Nevertheless, data from the current study raise new questions regarding the potential differences in the mechanisms of secondary injury related to Bace1 across the age spectrum of TBI and injury severity. The data also suggest that potential therapies targeting Bace1 may need to be tailored according to age, as their use may exacerbate functional deficits after TBI in younger patients
Footnotes
Acknowledgments
This work was supported by grants from the National Institute of Child Health & Human Development 5K12HD052896 (R.C.M.), the Charles Hood Foundation (R.C.M.), and the National Institute of Neurological Disorders and Stroke 5RO1NS047447 (M.J.W.).
Author Disclosure Statement
No competing financial interests exist.