
Abstract
Multiple lines of evidence have validated the Rho pathway as important in controlling the neuronal response to growth inhibitory proteins after central nervous system (CNS) injury. A drug called BA-210 (trademarked as Cethrin®) blocks activation of Rho and has shown promise in pre-clinical animal studies in being used to treat spinal cord injury (SCI). This is a report of a Phase I/IIa clinical study designed to test the safety and tolerability of the drug, and the neurological status of patients following the administration of a single dose of BA-210 applied during surgery following acute SCI. Patients with thoracic (T2-T12) or cervical (C4-T1) SCI were sequentially recruited for this dose-ranging (0.3 mg to 9 mg Cethrin), multi-center study of 48 patients with complete American Spinal Injury Association assessment (ASIA) A. Vital signs; clinical laboratory tests; computed tomography (CT) scans of the spine, head, and abdomen; magnetic resonance imaging (MRI) of the spine, and ASIA assessment were performed in the pre-study period and in follow-up periods out to 1 year after treatment. The treatment-emergent adverse events that were reported were typical for a population of acute SCI patients, and no serious adverse events were attributed to the drug. The pharmacokinetic analysis showed low levels of systemic exposure to the drug, and there was high inter-patient variability. Changes in ASIA motor scores from baseline were low across all dose groups in thoracic patients (1.8±5.1) and larger in cervical patients (18.6±19.3). The largest change in motor score was observed in the cervical patients treated with 3 mg of Cethrin in whom a 27.3±13.3 point improvement in ASIA motor score at 12 months was observed. Approximately 6% of thoracic patients converted from ASIA A to ASIA C or D compared to 31% of cervical patients and 66% for the 3-mg cervical cohort. Although the patient numbers are small, the observed motor recovery in this open-label trial suggests that BA-210 may increase neurological recovery after complete SCI. Further clinical trials with Cethrin in SCI patients are planned, to establish evidence of efficacy.
Introduction
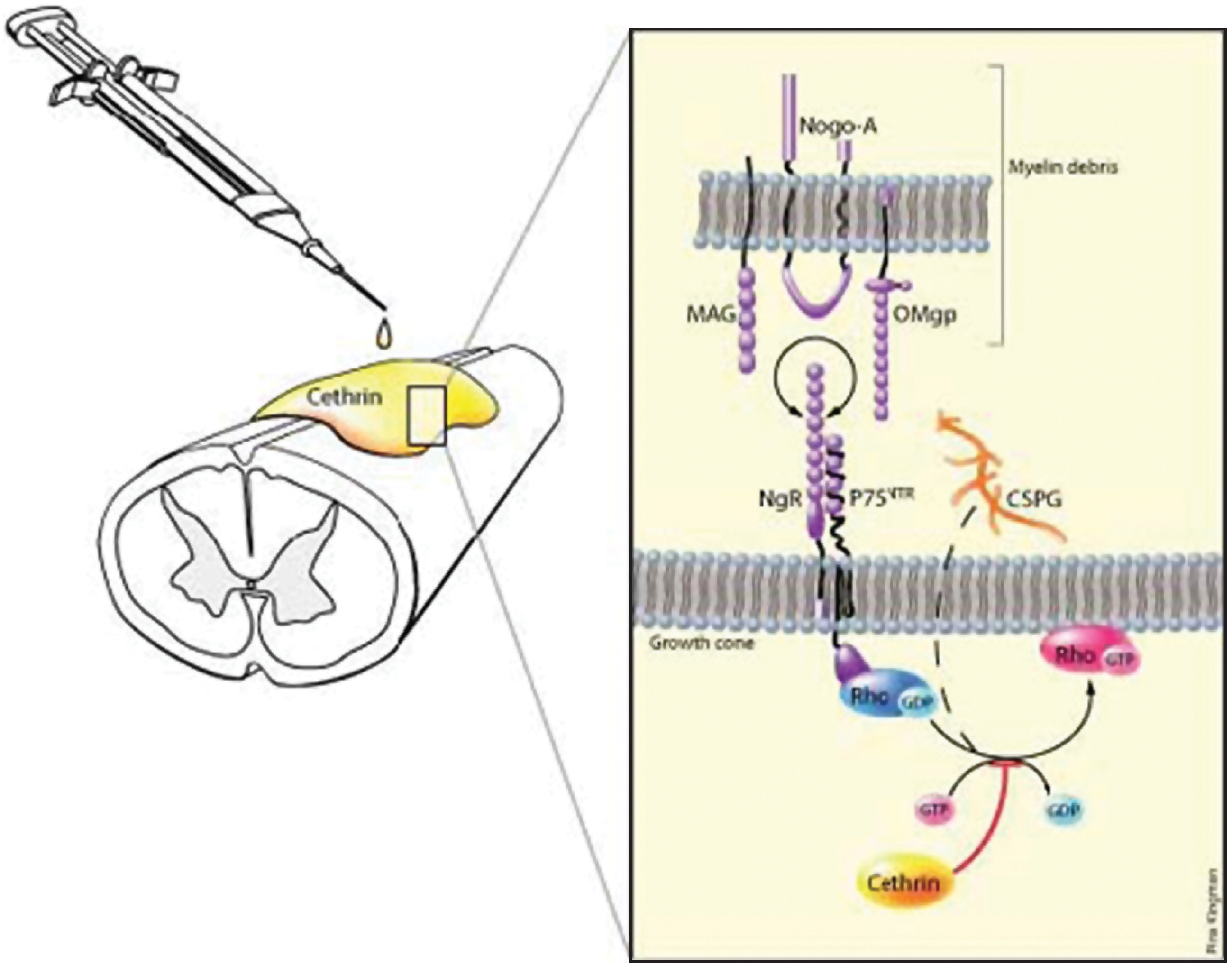
Schematic of Cethrin application and mechanism of action. Cethrin is applied to the injured spinal cord extradurally and exerts its effects by blocking the activation of Rho, thereby encouraging neurite outgrowth.
C3 transferase, an enzyme from Clostridium botulinum, blocks the Rho signalling pathway by locking RhoA in the inactive state. C3 transferase has been shown to reverse the effect of extracellular inhibitory substrates on neurons in culture and to enable the outgrowth of neurites (Jin and Strittmatter, 1997; Lehmann et al., 1999). Local application of C3 transferase to the injury site in a mouse hemi-section model of SCI resulted in long-distance regeneration of cortico-spinal neurons, increased expression of growth-associated protein-43 in the motor cortex, and improved functional recovery as assessed by locomotor scores and limb coordination (Dergham et al., 2002; Lord-Fontaine et al., 2008). However, cell penetration by the wild-type C3 transferase is very low, limiting its clinical development as a therapeutic agent. BA-210 is a recombinant engineered variant of C3 transferase that can readily cross the dura of the spinal cord as well as the cell membrane via a receptor-independent mechanism, and is being developed under the drug Cethrin (Lord-Fontaine et al., 2008). Epidural administration of Cethrin to the site of injury in a fibrin sealant carrier inactivates RhoA, reduces the extent of the lesion, and improves functional recovery in rodent models of SCI. Moreover, Cethrin penetrates to the ventral spinal cord when applied to the dorsal spinal cord in animal models (Lord-Fontaine et al., 2008).
This clinical trial was undertaken to assess the safety and tolerability of the drug, and the neurological status of patients treated with escalating doses Cethrin (ranging from 0.3 mg to 9 mg). We chose a noninvasive, fibrin-mediated delivery method, as its use has been supported by experimental studies in rodents (Guest et al., 1997; Kassam et al., 2004) and it is used routinely by spinal surgeons (Nakamura et al., 2005). We hypothesized that a single dose of Cethrin, applied to the dura mater of the injured spinal cord, would be well tolerated and clinically feasible to administer, and would improve the neurological outcome of patients with cervical or thoracic SCI as measured by the American Spinal Injury Association (ASIA) assessment.
Methods
Subjects and study design
The planned enrollment was 35–60 patients, and a total of 48 patients were enrolled in the trial, 32 with thoracic injury (T2-T12), and 16 with cervical injury (C4-T1) aged 16–70 years inclusive. All patients had acute, complete (ASIA Impairment Scale Grade A) SCI and were scheduled to undergo spinal decompression surgery or other interventional spinal surgery (e.g., fixation) within 7 days of injury. The study was approved as an Investigation of a New Drug by the Food and Drug Administration (USA) and by a Clinical Trial Application to Health Canada (Canada), and monitored by an independent Data-Safety Monitoring Board (DSMB). Age-eligible participants were recruited from February 3, 2005 to November 15, 2007. Patients were selected for the study from nine clinical sites in the United States and Canada.
Five different doses of Cethrin were studied; 0.3mg, 1mg, 3mg, 6mg, and 9mg. Dosing was first tested in thoracic patients because this cohort would be least affected if there was worsening in the level of neurological injury, as recommended by the International Campaign for Cures for spinal cord injury Paralysis (ICCP) guidelines (Fawcett et al., 2007). After the DMSB determined that the drug was safe in at least five thoracic patients (Figure 2), cervical patients were enrolled at the same dose. The DMSB also reviewed the safety data before approval of dose escalation. Each patient received a single application of Cethrin at the time of surgery. The route of administration was a local application of Cethrin onto the anterior or posterior dural surface area overlying the injured spinal cord, upon completion of decompression and instrumentation, for patients undergoing spinal decompression or fixation surgery by a neurosurgeon or orthopaedic surgeon. The protocol directed a solitary application of Cethrin for each patient.
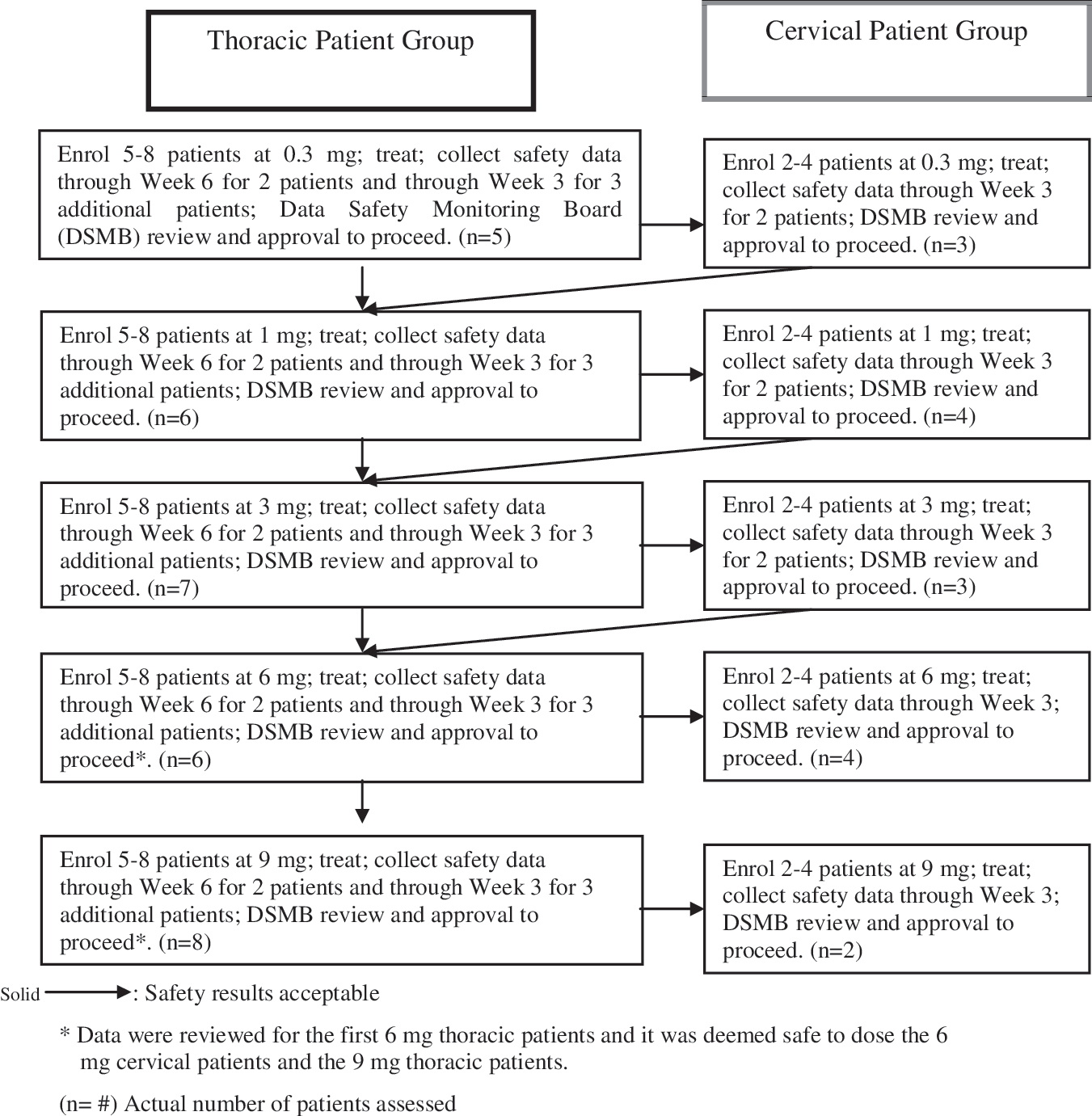
Study design for patient recruitment.
Patients were assessed for suitability for entry into the trial during the pre-study period: vital signs, physical examination, clinical laboratory tests, and ASIA assessment were performed. All centers in the study adhered to the acute SCI management guidelines published by a consensus panel in 2002 (Hadley et al., 2002). These guidelines included avoidance of hypotension with induced hypertension (mean>85 mm Hg), and early intervention with surgical decompression and reconstruction (within 72 h when medically feasible). The mean time for drug administration was at 52 h as described in detail in the Results section. Baseline computed tomography (CT) scans of the spine, head, and abdomen were completed during the pre-study period. Magnetic resonance imaging (MRI) of the spine in the area of presenting injury and of the head were preformed during the pre-study period, 72–96 h following surgery, and at week 6. Results of any additional scans performed at the investigator's discretion as part of the standard of care were recorded.
Inclusion/exclusion criteria
Potential participants were excluded if any of the following conditions were present: (1) use of any experimental drug, or participation in any clinical trial, within 30 days prior to surgery; (2) history of adverse reaction to fibrin sealant; (3) history of hypersensitivity to bovine products; (4) any medical condition that might interfere with the ASIA assessments; (5) clinically significant neurological, cardiac, respiratory, hepatic, or renal disease or malignancy; (6) hemophilia or other bleeding abnormality as defined by a platelet level<100×109/L, activated partial thromboplastin time or international normalized ratio higher than the upper limit of normal, and/or a baseline hematocrit <0.25; (7) gunshot wound as the presenting injury, or any evidence of transecting injury to the spinal cord (e.g., by stab wound); (8) cognitive impairment that could preclude accurate neurological assessments (e.g. traumatic brain injury with Glasgow Coma Scale of ≤14); (9) ankylosing spondylitis; (10) diabetes mellitus requiring insulin therapy; (11) known immunodeficiency, including AIDS, or use of immunosuppressive or cancer chemotherapeutic drugs; (12) pregnancy or breastfeeding during the study; (13) any condition or situation likely to cause the patient to be unable or unwilling to participate in the study or follow-up; (14) any condition likely to result in the patient's death within the next 6 months; (15) any other condition that, in the opinion of the investigator, would preclude the patient's participation in the study; (16) previous participation in this study; (17) use of intravenous heparin in the previous 48 h or thrombolytics and/or aspirin containing products in the previous 10 days; or (18) non-traumatic causes of spinal cord injury (e.g. transverse myelitis, acute disc herniation). Dural tear was not a contraindication to enrolment. Of the recuited patients 31% were reported to have dural tears, and in 69 % the dura was intact.
Dose and drug delivery
The starting dose of 0.3 mg chosen for study was based on the “no observed adverse effect” level determined from pre-clinical safety studies. A total of seven toxicology studies, two non-Good Laboratory Practice (GLP) studies and five GLP studies, were conducted in rats, dogs, and pigs to evaluate the safety of Cethrin administered intravenously and subcutaneously, as well as Cethrin prepared with Tisseel® administered extradurally to the spinal cord. The five GLP toxicology studies comprised four acute studies in rats and dogs and one repeated-dose study in rats. In these GLP studies, more than 400 rats and 24 dogs were studied. The human equivalent safe doses were calculated to be 3.5 mg/kg based on body weight, or 10 mg based on relative spinal cord surface area. The starting dose of 0.3 mg in humans was predicted to be within the effective dose range, based on contusion studies in rats, and Rho inactivation patterns in injured rat spinal cord after application of BA-210 (Lord-Fontaine et al., 2008).
For effective and simple delivery of Cethrin to the site of SCI, BA-210 was combined with a fibrin sealant product at the clinical site (Fig. 1). Fibrin sealants are provided as single-use kits consisting of four components (five components in Canada) packed in separate vials (Tisseel; Baxter, Deerfield, IL). The 1 mL size was used in this study. The components of the kit are fibrinogen and reconstitution buffer containing bovine aprotinin; thrombin (in Canada two thrombin vials are included and 500 IU/mL was used) with reconstitution buffer containing calcium chloride. The fibrinogen and thrombin were reconstituted by mixing with their respective buffer solutions. BA-210 was formulated in 5 mM sodium citrate buffer pH 6.5 and was supplied as 0.5 mL in a 2 mL vial at a concentration of 30 mg/mL. The BA-210 solution was kept frozen at −20°C until used. The final BA-210 doses were prepared within 1 h prior to surgery from the 30 mg/mL solution and co-applied with the thrombin and fibrinogen solutions by use of the Duploject® applicator (Baxter Healthcare Corporation, Deerfield, IL). The applicator is a double-barreled syringe that separates the fibrinogen and thrombin until the plunger is pushed, which mixes the solutions simultaneously with delivery.
Outcome measures
The primary objective was to determine the safety and tolerability of escalating doses of BA-210. The primary safety parameter was unexpected worsening in neurological function at week 6. The secondary objectives were to evaluate the pharmacokinetic profile of BA-210, immunologic parameters following exposure to BA-210, and the neurological status of patients as measured by the ASIA assessment (Steeves et al., 2007). The motor score ASIA assessments were completed within 12 h prior to surgery and between 48 and 72 h following surgery, as well as at postoperative months 3, 6, and 12 as follow-up. Safety was assessed by evaluating vital signs, clinical laboratory and MRI results, as well as any adverse events. All patients received MRI scans of the brain and spinal cord prior to surgery, as well as 3 days and 6 weeks after BA-210 application, to assess the safety of the treatment and to exclude the possibility of exacerbation of the lesion by the experimental treatment.
Safety and adverse events
Adverse events were recorded as follows: reports spontaneously volunteered by the patients; events recorded by the investigator; and investigator-elicited reports in response to open-ended questions. Patients with acute thoracic (T2-T12) SCI were treated before patients with acute cervical (C4-T1) SCI and cervical patients were only recruited into the study once there was cumulative safety data for at least two patients from the same dose group who had completed the week 6 assessments (Fig. 2).
The dose level of a patient was determined by the dose the patient received, not necessarily the dose to which he or she was assigned. Three deviations were reported for dose administration: (1) Patient 01-1-02 was assigned to receive 0.3 mg of BA-210. Analysis of the dosing solution revealed that the patient had received a 3-mg dose of BA-210. (2) Patient 04-1-01 was assigned to receive 1 mg of BA-210. The patient had received a 2-mg dose of BA-101. (3) For Patient 08-1-02, the time that the dose was administered was not recorded; therefore, nominal times of sample collections were used to compute parameters.
All patients who were enrolled and received one dose of study medication were included in the summaries and analyses for safety, including any patient who withdrew early from the study. A DSMB periodically reviewed safety data from the study and made recommendations to stop/continue enrollment of patients at a particular dose level or stop/continue the study. The primary population for efficacy analysis was the intent-to-treat population that included all patients who were enrolled and received any dose of study medication. Treatment-emergent adverse events were defined as all adverse events occurring during the study period.
Pharmacokinetic analysis
Blood samples were obtained for pharmacokinetic assessments before surgery and at 0.5, 1, 2, 6, 24, and 48 h following Cethrin application [during the intensive care unit (ICU) period]. Serum concentrations of BA-210 were determined by enzyme-linked immunosorbent assay (ELISA).
Antibody levels
Blood samples were obtained to measure antibody levels in serum before surgery and on day 10, week 6, and month 6 assessments. An ELISA assay was used to measure serum concentrations of BA-210.
Neurological outcomes
Changes in ASIA grade from baseline to each of the planned ASIA grade times for total motor score were summarized using the mean and standard deviation. The number and percentage of patients who shifted from category A on the ASIA impairment scale to categories B, C, D, or E were tabulated by patient group, dose level, and time for observed data.
Results
We enrolled 48 patients with acute, complete thoracic, or cervical SCI (ASIA grade A) and thoracic patients were treated and assessed at each dose before the recruitment of cervical patients (Fig. 2). Patients received a single dose of BA-210 in fibrin 7.83 to 146.1 h after the initial injury (mean 52.6 h). Thirty-five of the 48 patients enrolled completed the trial: 23 with complete thoracic SCI, and 12 with complete cervical SCI. Of the patients who did not complete the study, six were lost to follow-up, three were lost during the company transition, two died, one withdrew consent, and one refused to return to complete visits. The patients in the cervical and thoracic groups were well matched for demographics, age, and gender (Table 1).
Patient 01-1-02 received a 3-mg dose instead of the assigned 0.3-mg dose; patient included according to the actual dose.
Patient 04-1-01 received a 2-mg dose instead of the assigned 1-mg dose; patient included according to intended dose.
Adverse events
The primary objective of the trial was to measure the safety and tolerability of Cethrin when administered in conjunction with fibrin sealant to the dura mater of the spinal cord. Safety was assessed by vital signs, physical examination, clinical laboratory and MRI results, as well as adverse events. Neurological function was assessed by the ASIA assessment. The adverse event profile indicated that Cethrin is safe and tolerable in the dose range studied (0.3–9 mg). The reported treatment-emergent adverse events were typical for a population of acute SCI patients, and there were no serious adverse events that were attributed to Cethrin by the clinical investigators and the DSMB. In particular, there were no allergic reactions, cardiopulmonary complications, or hemorrhagic adverse events attributable to the medication. Table 2 summarizes the serious adverse events in both the thoracic and cervical cohorts in this trial. Two subjects died while enrolled in the trial: one from adult respiratory distress syndrome; one in a delayed fashion from a cerebral glioblastoma that became apparent after enrolment in the trial. These deaths were attributed to causes related to the subjects' initial SCI, other injuries, or pre-existing conditions, and were within or below the expected death rate for an acute SCI population (Furlan et al., 2010; Geisler et al., 2001) and determined not to have a causal relation to Cethrin. The overall mortality was 2/48 patients (4%).
The urinary tract infection rate (a subset of infections, Table 2) was consistent at all dose levels for the thoracic cohort of patients (mean 75%); however, in the cervical cohort of patients the urinary tract infection rate was lower, (mean 50%), and none of the patients in the 3-mg group had urinary tract infection.
Pharmacokinetics
A summary of pharmacokinetic parameters is shown in Table 3. For all treatment groups, the peak concentration (CMAX) and the area under the curve parameters exhibited high inter-patient variability. There was almost a twofold greater observed maximum concentration when the dose was applied to the thoracic site compared to the cervical site of SCI for the 3-mg and 6-mg doses. The highest serum concentration of the drug was in patients treated with 9-mg of Cethrin. The mean Cmax value at the effective dose range (1–3 mg) was 0.8 to 2.3 for the cervical and thoracic patients, respectively. This value is below the mean Cmax value (3.5 ng/mL) associated with a dose of 0.01 mg in rats in the extradural toxicity study, which was considered a “no observed adverse effect” level. In addition, the mean Cmax and AUC values for all of the patients in this study were below the Cmax and AUC values for the low doses tested in the intravenous toxicity studies in rats and dogs.
NA, not available.
Number of patients for the T1/2 values were n=1, n=6, n=3, n=3 for 1-mg, 3-mg, 6-mg, and 9-mg values, respectively, and were n=3 for the 3-mg cervical group.
In cervical patients, only those treated with doses of ≥3 mg had levels of the drug that were above the limit of detection of 100 pg/mL. At all doses except the 9-mg dose, the plasma levels were higher in the thoracic SCI patients than in the cervical patients. At the very lowest doses, very few patients had measureable concentrations of Cethrin. All of the samples from patients with cervical SCI who received the 0.3-mg and 1-mg dose were below the limit of quantitation
Antibody testing
Cethrin is a protein drug and patients were tested for the development of antibodies against the drug at day 10, week 3, week 6, month 3, and month 6. By month 6, 50% of patients in the thoracic group and 38% of patients in the cervical group had developed detectable levels of anti-Cethrin antibody.
Neurological outcomes
Recovery of motor function in patients with complete SCI is fairly limited, and therefore, neurological motor scores (ASIA Impairment Scale motor scores) are often utilized in Phase I and Phase II SCI trials to determine if an experimental intervention has potential beneficial effects (Fawcett et al., 2007). We observed a large preliminary effect in ASIA motor scores in subjects with complete cervical SCI. The largest mean changes in ASIA motor scores were seen in the cervical patients treated with 1 mg and 3 mg of Cethrin (Fig. 3). The ICCP Clinical Guidelines Panel reports that a cervical ASIA Grade A-injured patient is likely to spontaneously improve by approximately 10 motor points during the first year after SCI, whereas we observed average improvements of 21.3 and 27.3 points in the 1-mg and 3-mg groups, respectively, at 12 months (Fig. 4). There was little, if any, effect in the thoracic injury cohort, and the average change for baseline for all doses in thoracic patients was (1.8±5.1) compared to an overall average change of (18.6±19.3) for all cervical patients. Analogous to the findings with motor scores, patients in the cervical cohort showed improvement in sensory scores. Very little, if any, improvements in sensory scores were observed in the thoracic cohort, at any dose (data not shown).
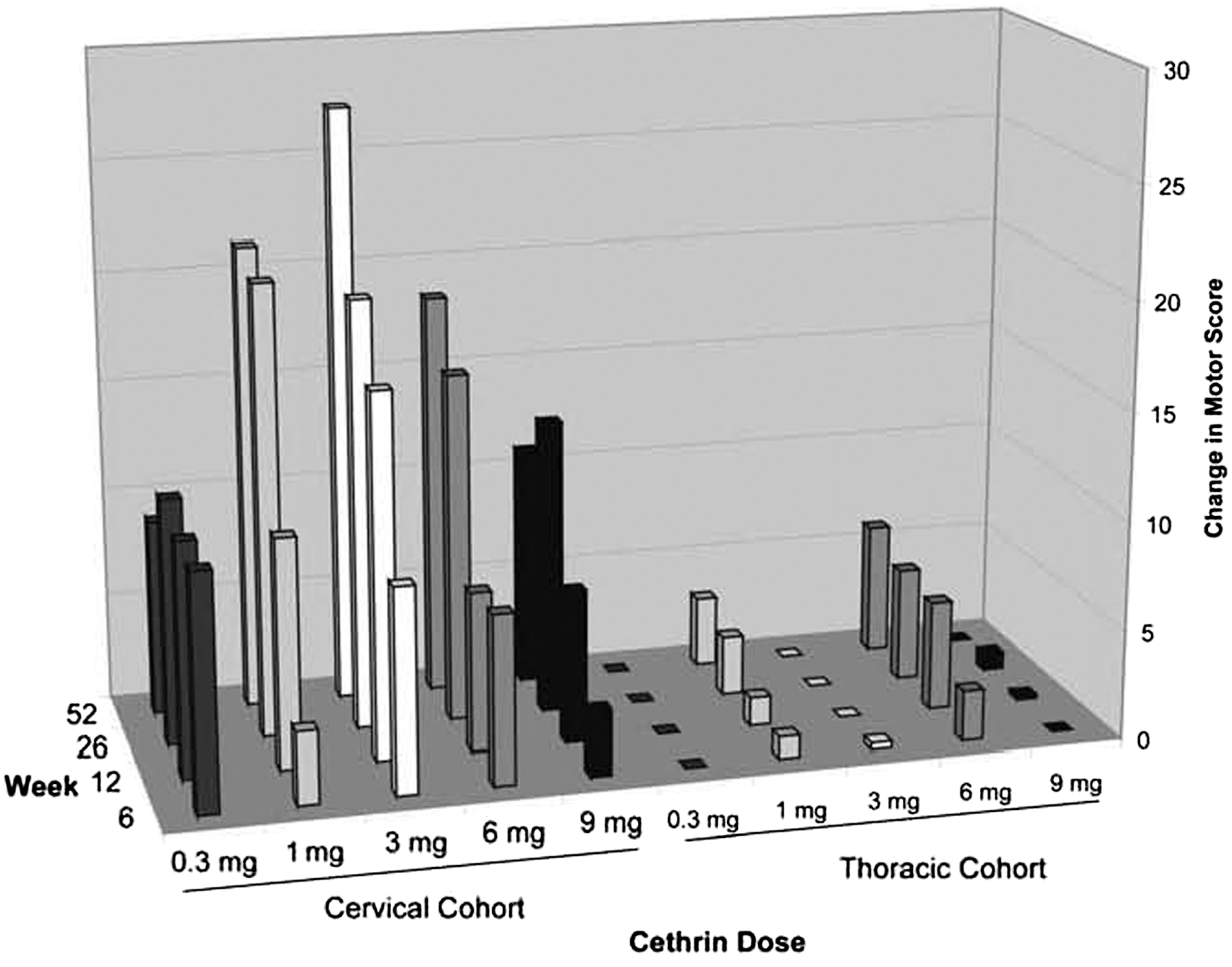
Change in motor score over time for all treatment. The cervial cohort of patients and thorcic cohort of patients were followed for 52 weeks. The mean change in ASIA motor scores is shown for each dose group at each time point. Ten key muscle groups, five in the arm and five in the leg, were tested bilaterally, and given a score of 0–5 each, resulting in a possible total score of 100.
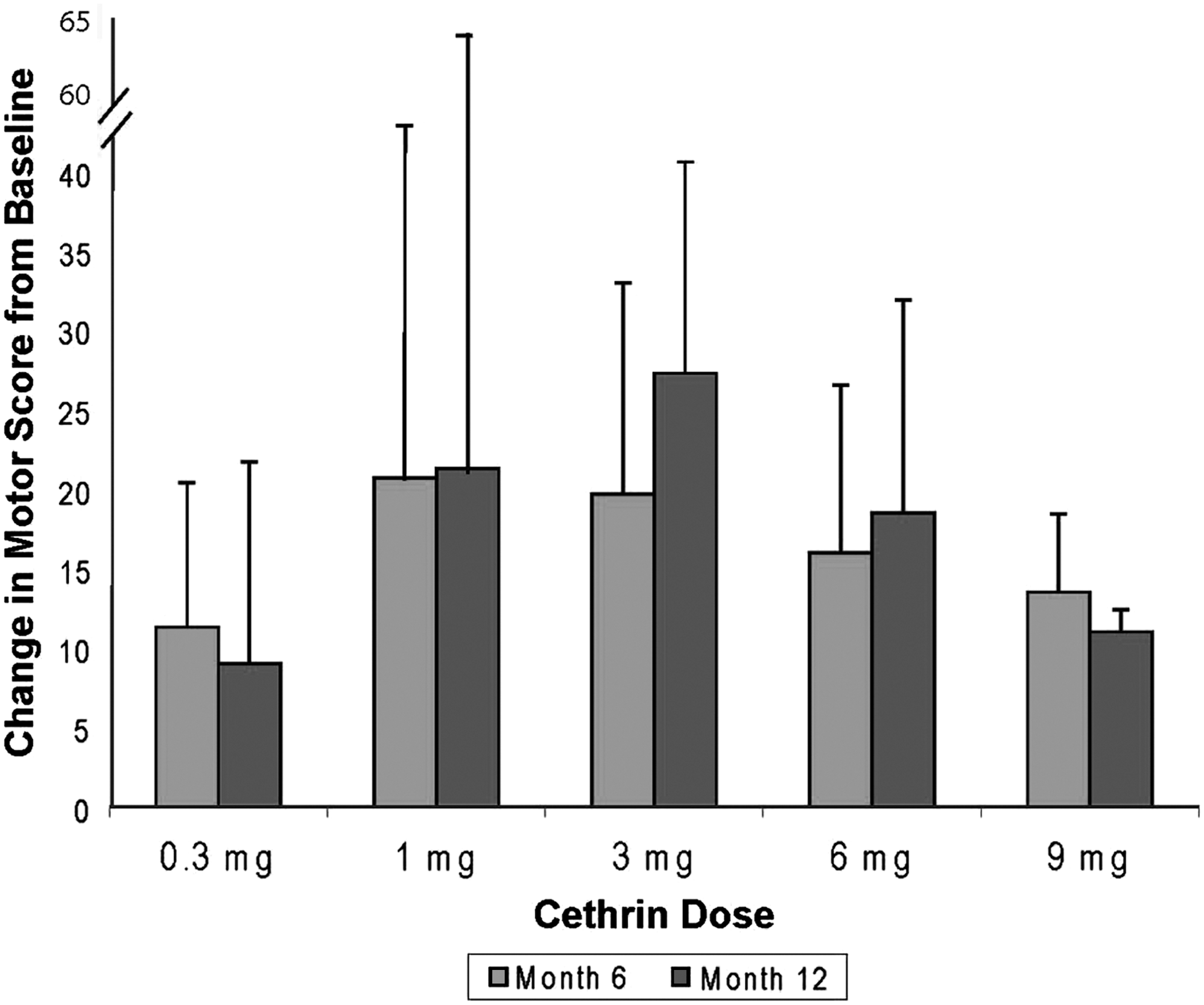
Change in motor score for cervical patients at 6 and 12 months. The average change in motor score±standard deviation for the cervical patients is shown at 6 and 12 months.
Motor function data were also analyzed in terms of conversion on the ASIA Impairment Scale. All patients recruited into the study had a severe SCI, and were classified as ASIA A. At 12 months after treatment 31% (5/16) of subjects with cervical SCI and 6.3% (2/32) of subjects with thoracic SCI improved over the time period to at least ASIA C. In the 3-mg cervical group, 66% improved to ASIA C or better. One cervical SCI subject in each of the 1-mg and 3-mg dose cohorts improved to ASIA Grade D (Fig. 5), and one subject with a thoracic injury in the 6-mg dose cohort improved to ASIA Grade D.
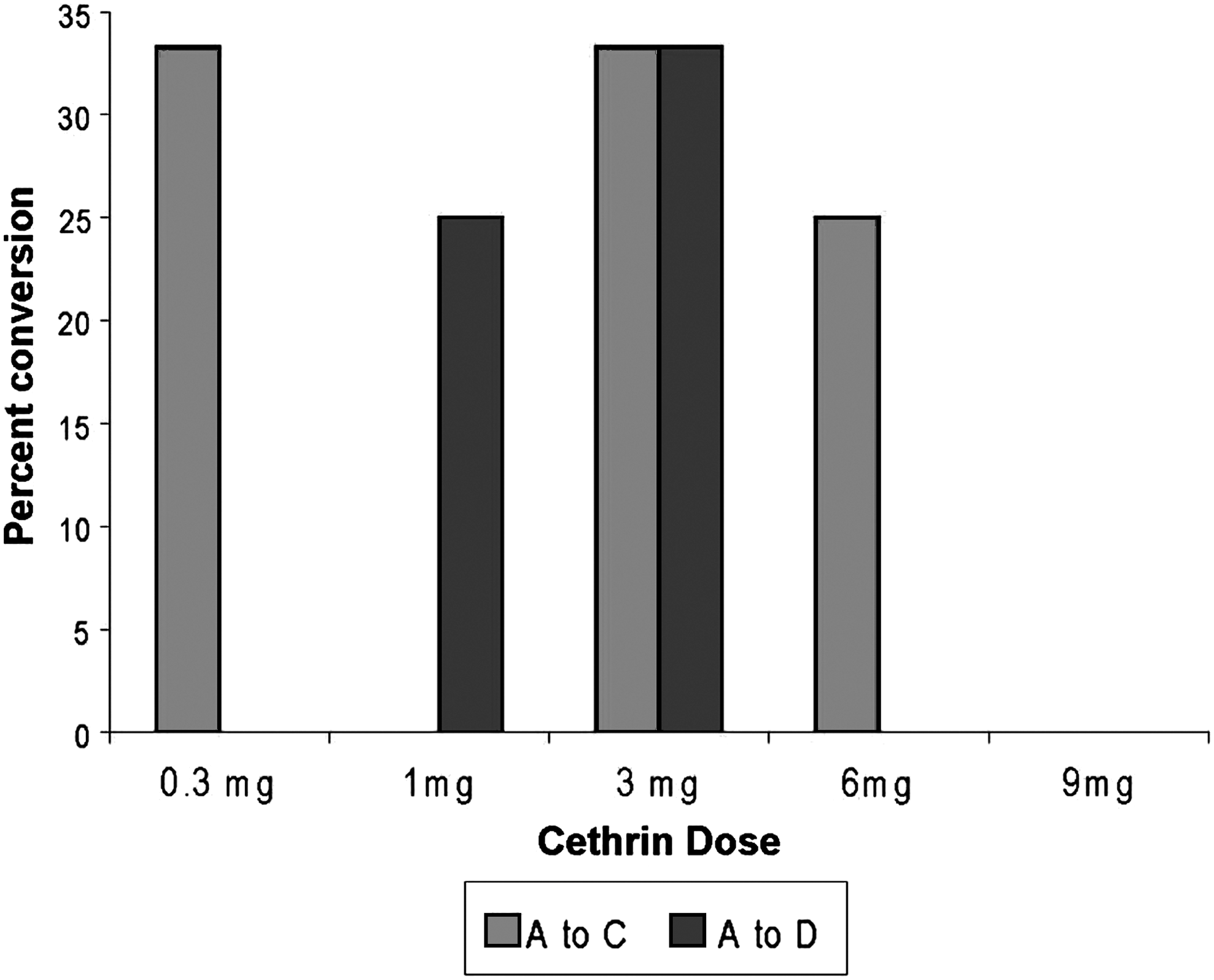
Conversion of cervical patients. Percent of patients that converted from ASIA A to ASIA C (light grey) or to ASIA D (dark grey) is shown for all of the dose groups.
Discussion
In this multicenter trial involving nine sites, we report for the first time a safe and effective method of delivering a biological drug to the injured spinal cord that does not confer the risks associated with direct injection in the spinal cord or intrathecal administration, techniques that have been used in other SCI clinical trials (Blight and Tuszynski, 2006). The primary outcome measure for this trial was the safety and tolerability of increasing doses of Cethrin. Adverse events for both the thoracic and cervical cohorts were most frequently recorded for the following system organ classes: gastrointestinal disorders; general disorders; infections; and psychiatric disorders. None of the serious adverse events were assessed as drug related. The incidence of events was consistent across dose levels and was attributed to the original injury or associated with medication administered to alleviate conditions that stemmed from the original injury, such as constipation, nausea, urinary tract infection, anxiety, depression, and insomnia. It should be noted that adverse events are expected to occur in patients following SCI.
MRI was used during the pre-screening period, at 72–96 h following surgery, and at week 6 as a qualitative measure to detect the location of the injury, degree of severity, and potential complications. Whereas MRI was an essential diagnostic tool in this study, it does not as yet have the resolution to be used to follow recovery or predict outcome.
Two patients died during this study, which is in the range or lower than the expected number of deaths in an acute SCI population (Furlan et al., 2010; Giester et al., 2001). One of these patients, a 67- year-old white man with a thoracic SCI, died of acute respiratory distress syndrome (ARDS) 18 days following injury. A second patient, a 52-year-old woman with a thoracic SCI, died of a cerebral malignant astrocytoma 11 months after injury. Subtle evidence of cerebral lesion was seen on the admission brain CT on retrospective review although it was not appreciated at the time of study entry.
Thoracic patients were included in the study primarily to assess the safety of Cethrin. Thoracic patients were slow to recruit because many patients with thoracic SCI often have injuries that encompass a wide range of tissues and therefore did not meet the inclusion/exclusion requirements. Thoracic injuries are often associated with significantly more trauma to surrounding tissues than are cervical injuries, creating an environment that is more likely to allow for systemic absorption of the locally-administered Cethrin and potentially greater systemic toxicity. Indeed, in the Phase I/IIa trial, thoracic patients had approximately twice the blood levels of Cethrin at a given dose than did cervical patients, except at the 9-mg dose.
The pharmacokinetic analysis, a secondary outcome measure, indicated that there was little systemic exposure to the drug in patients. The mean Cmax value at the dose range of 1–3 mg for the cervical and thoracic patients was 0.8-2.3 ng/mL, respectively, and was below the mean Cmax value for the “no observed adverse effect” level determined from an extradural toxicity study in rats. Moreover, the Cmax and AUC values for the all of the patients in this study were below the Cmax and AUC values for the low doses tested in the intravenous toxicity studies in rats and dogs. At all doses except the 9-mg dose, the plasma levels were higher in the thoracic SCI patients than in the cervical patients. This discrepancy is probably because the thoracic spine is a much more stable structure than the cervical spine, and a much more significant trauma is required for a severe (ASIA A) injury of the thoracic spinal cord. The greater traumatic disruption of the tissues in the thoracic SCI patients increases the chance of systemic exposure to an epidural injection. Variation in patient weight could also contribute to variable systemic exposure.
It is reported that >50% of people living with SCI have some episodes of chronic neuropathic pain. Causing pain as part of a treatment to promote regeneration of injured fibers is a concern because some experimental treatments tested in animals have been shown to stimulate fiber growth in central pain pathways. However, pre-clinical studies in rats indicated that the inactivation of Rho signalling would have no effect on, or would even decrease, pain (Lord-Fontaine et al., 2008; Ramer et al., 2004). In this clinical study pain was reported as a treatment-emergent adverse event; there was no evidence that Cethrin affected pain, either positively or negatively.
Although this Phase I/IIa trial was open label and the analysis was not blinded, the neurological outcomes are encouraging based on both ASIA motor scales in patients with cervical injury, and the conversion rate from ASIA A to ASIA C or D. We observed conversions to ASIA C or D in 31% of patients with cervical SCI and in 6% of thoracic SCI patients. When pooled, the conversion rates are typically higher in cervical patients, and the predicted rate of conversion in a untreated cervical SCI population would be ≤10% (Fawcett et al., 2007). The observed change in cervical, but not thoracic, patients is expected based on the anatomy of the spinal cord, motor innervation patterns, and their relative contribution of these differences to ASIA scoring (Fawcett et al., 2007). Whereas much emphasis is typically placed on motor recovery, the fact that the cervical patients achieved substantial sensory recovery is also noteworthy, as such sensation may have a crucial protective function for skin areas that are sensitive to pressure sores, and the sensory feedback may help in seating balance (Anderson, 2004).
There is less spontaneous recovery in ASIA A patients than in patients with incomplete injury, and based on a number of published studies, recovery in ASIA A patients is quite predictable. In the first year after SCI, an ASIA A patient is expected to improve ∼10 ASIA motor points (Steeves et al., 2007). Although we did not have a placebo arm in this safety trial, the improvement in the cervical patients of 21.3 and 27.3 in the 1-mg and 3-mg dose groups, respectively, is very encouraging. However, it should be noted that many patients living with SCI show some recovery after the first few days under intensive care, and it has been argued that an ASIA assessment at 24 h is not as reliable as one taken at 72 h when the patient is more stable (Steeves et al., 2007). The continued improvement that we observed between week 6 and week 52 (Fig. 3) is also encouraging. If some recovery were caused by regeneration of injured fibers, this would be expected to be a slow process, similar to what was observed.
At present, limited treatment options are available to patients with SCI, and the findings reported here on the safety and tolerability of Cethrin indicate a potential new drug for further study to treat acute SCI. Targeting Rho to foster both neuroprotection and axon regeneration is well understood in terms of mechanism of action, and a wealth of data from animal studies provides good rationale for a progression to clinical studies. We show here encouraging findings with a small number of patients that indicate some evidence of efficacy to enhance functional recovery. Further placebo-controlled clinical trials in SCI with Cethrin will further our understanding of the effects of this intervention on neurological recovery patterns and functional improvement in daily activities. We hope that the success of this Phase I/IIa trial will provide encouragement for the translation of other promising therapies to treat SCI.
Footnotes
Acknowledgments
This trial was funded by BioAxone Therapeutics and Alseres Pharmaceuticals. The authors thank M. R. Hurtt, S. M. Flint, and B. A. Haynes of Alseres Pharmaceuticals for their thoughtful contributions and critical editing of this manuscript.
Author Disclosure Statement
Lisa McKerracher is a co-inventor of Cethrin and has proprietary interests in the compound. Michael Fehlings was previously a member of the Scientific Advisory Board of Bioaxone and Alseres Pharmaceuticals.